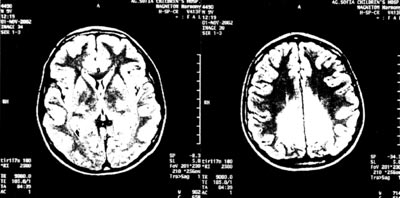
Εικόνα 1.
ΜRΙ εγκεφάλου.
Παθολογικό σήμα σε οπίσθια περικοιλιακή λευκή ουσία.
Φυλοσύνδετη Αδρενολευκοδυστροφία:
περιγραφή περίπτωσης
Α. Παπαλουκάς
Δ. Λαζοπούλου
Σ. Γιουρούκος
Ε. Δρογκάρη
Γ. Χρούσος
Α΄ Παιδιατρική Κλινική
Πανεπιστημίου Αθηνών, Νοσοκομείο Παίδων "Αγία Σοφία"
Υποβλήθηκε:
18/9/2003
ΠΕΡΙΛΗΨΗ
Η αδρενολευκοδυστροφία αποτελεί νοσολογική οντότητα η οποία ανήκει στις διαταραχές
της λειτουργίας των υπεροξεισωματίων και κληρονομείται με το φυλοσύνδετο υπολειπόμενο
χαρακτήρα. Περιγράφεται περίπτωση αδρενολευκοδυστροφίας σε αγόρι ηλικίας 9,5
ετών, το οποίο νοσηλεύθηκε στην κλινική μας για διερεύνηση επεισοδίου σπασμών.
Η διάγνωση πιθανολογήθηκε από την κλινική εικόνα και τη μαγνητική τομογραφία
εγκεφάλου, στην οποία διαπιστώθηκαν παθολογικά ευρήματα συμβατά με τη νόσο,
και τέθηκε με την ανεύρεση στο πλάσμα υψηλής συγκέντρωσης λιπαρών οξέων πολύ
μακράς αλύσου (VLCFA). (Δελτ Α΄ Παιδιατρ Κλιν Πανεπ Αθηνών 2004, 51(1):58-65)
Λέξεις ευρετηριασμού: φυλοσύνδετη αδρενολευκοδυστροφία, VLCFA, μεταμόσχευση μυελού οστών, Lorentzo's oil.
ΕΙΣΑΓΩΓΗ
Η φυλοσύνδετη αδρενολευκοδυστροφία (Χ-linked ALD) ανήκει στην ευρύτερη ομάδα
των διαταραχών της λειτουργίας των υπεροξεισωματίων.[1,2] Κληρονομείται με το
φυλοσύνδετο υπολειπόμενο χαρακτήρα και έχει παγκόσμια κατανομή ανεξάρτητα φυλής,
ενώ η συχνότητά της ανέρχεται σε 1/20.000-1/50.000 άτομα στο γενικό πληθυσμό,
ή 1/100.000 άρρενες γεννήσεις. Η θέση του γονιδίου το οποίο είναι υπεύθυνο για
τη νόσο, εντοπίζεται στο μακρύ σκέλος του χρωμοσώματος Χ στη θέση 28 (Χq28)
και έχουν διαπιστωθεί πάνω από 300 διαφορετικές μεταλλάξεις.
Η βιοχημική ανωμαλία "κλειδί" της νόσου είναι η συσσώρευση λιπαρών
οξέων πολύ μακράς αλύσου -very long chain fatty acids: VLCFA (άτομα άνθρακα:
C >22)- στους ιστούς, και κυρίως στη λευκή ουσία του εγκεφάλου, στο φλοιό
των επινεφριδίων και στους όρχεις. Η βιοχημική και μοριακή διεργασία της νόσου
είναι πολύπλοκη, πολυενζυμική και σε πολλά σημεία, ακόμη και σήμερα, άγνωστη.
Αξίζει να τονισθεί ότι, ο γονότυπος δεν καθορίζει το φαινότυπο της νόσου.[3,4,5]
Το γεγονός αυτό αρχικά είχε αποδοθεί στην πιθανή ύπαρξη κάποιου τροποποιητικού
γονιδίου. Μετά την παρουσίαση 3 ζευγαριών μονοζυγωτικών διδύμων με διαφορετικό
φαινότυπο, έγινε πιο πιθανή η υπόθεση ότι περιβαλλοντικοί παράγοντες είναι εκείνοι
που καθορίζουν το φαινότυπο της νόσου.
Η διάγνωση της Χ-ALD τίθεται με τη μέτρηση των VLCFA στο πλάσμα κυρίως, τα οποία
ανευρίσκονται αυξημένα.[8,9,10,11] Επίσης, τα VLCFA μετρούνται και στα ερυθροκύτταρα
και σε καλλιέργειες ινοβλαστών του δέρματος. Αυτά, σε συνδυασμό με τη μαγνητική
τομογραφία του εγκεφάλου, θέτουν τη διάγνωση της νόσου. Η συνηθέστερη αρχική
εντόπιση στην απεικόνιση του εγκεφάλου αφορά σε συμμετρικές περιοχές παθολογικού
σήματος στη λευκή ουσία γύρω από τις κοιλίες, τους οπίσθιους βρεγματικούς και
ινιακούς λοβούς. Παράλληλα, η διάγνωση της νόσου συμπληρώνεται και ενισχύεται
από τον ενδοκρινολογικό έλεγχο (λειτουργία επινεφριδίων και άλλες ορμόνες),
τη γενετική και γονιδιακή μελέτη και διάγνωση, τη νευροφυσιολογική και νευροψυχολογική
εκτίμηση (ακουολογικός, οφθαλμολογικός έλεγχος και έλεγχος νοημοσύνης) και βεβαίως,
τέλος, την κλινική εικόνα.
Εικόνα 1.
ΜRΙ εγκεφάλου.
Παθολογικό σήμα σε οπίσθια περικοιλιακή λευκή ουσία.
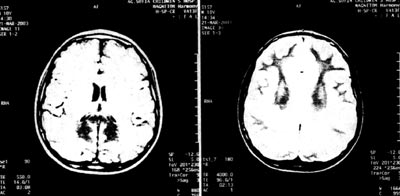
Εικόνα 2.
ΜRΙ εγκεφάλου.
Επιδείνωση αρχικών ευρημάτων και κατάληψη πρόσθιων περιοχών.

ΠΕΡΙΓΡΑΦΗ
ΠΕΡΙΠΤΩΣΗΣ
Αγόρι ηλικίας 9,5 ετών νοσηλεύθηκε στην κλινική μας τον Οκτώβριο του 2002, παραπεμπόμενο
από επαρχιακό νοσοκομείο, για διερεύνηση επεισοδίου εστιακών σπασμών με δευτερογενή
γενίκευση. Κατά τη διερεύνησή του στο εν λόγω νοσοκομείο, ετέθη η υποψία της
Χ-ALD με βάση τα ευρήματα της μαγνητικής τομογραφίας εγκεφάλου, όπου παρατηρήθηκε
παθολογικό σήμα στο έξω τριτημόριο των εγκεφαλικών σκελών, στο σπληνίο του μεσολοβίου
και στη λευκή ουσία παρά τα ινιακά κέρατα των πλαγίων κοιλιών. Επίσης, στο ηλεκτροεγκεφαλογράφημα
κατεγράφησαν υπό μορφή βραδυαρρυθμίας εστιακά ευρήματα στη δεξιά κρoταφοβρεγματική
περιοχή.
Το αγόρι είναι το πρώτο από τα τρία παιδιά, φαινοτυπικά υγιών γονέων, ενώ στην
οικογένεια υπάρχει και άλλος ένας μεγαλύτερος, ετεροθαλής αδελφός (από άλλο
πατέρα). Γεννήθηκε μετά από τελειόμηνο κύηση, με φυσιολογικό τοκετό. Η περιγεννητική
περίοδος ήταν ομαλή, όπως και η ψυχοκινητική του εξέλιξη, ενώ δεν αναφέρθηκαν
άλλες νόσοι, νοσηλείες ή επεισόδια σπασμών στο παρελθόν. Τέλος, όσον αφορά στο
οικογενειακό ιστορικό του ασθενούς, αρχικά αναφέρθηκε μόνο ότι ο μικρότερος
αδελφός παρουσιάζει περιστασιακά εμετούς σε περιπτώσεις συγκινησιακής φόρτισης
και ότι η μητέρα είναι ετερόζυγος β-μεσογειακής αναιμίας. Αναλύοντας καλύτερα
και επιμένοντας περισσότερο στο οικογενειακό ιστορικό του ασθενούς, προέκυψε
ότι και άλλα μέλη, άρρενα και θήλεα, από την οικογένεια της μητέρας, παρουσίαζαν
συμπτωματολογία συμβατή με τη νόσο, εκ των οποίων, συγκεκριμένα η αδελφή της
μητέρας επιβεβαιώθηκε ότι είναι ετερόζυγος Χ-ALD.
Η αντικειμενική εξέταση κατά την εισαγωγή του ασθενούς δεν παρουσίαζε παθολογικά
ευρήματα και η νευρολογική εξέταση ήταν φυσιολογική. Η αρτηριακή πίεση του αγοριού
ήταν εντός φυσιολογικών ορίων (114/61mmHg), όπως και οι σφύξεις του (90/min).
Ο εργαστηριακός έλεγχος, με γενική αίματος, βιοχημικό έλεγχο ορού και γενική
ούρων, που ελήφθη κατά την εισαγωγή του παιδιού, επίσης δεν κατέδειξε κάτι το
παθολογικό.
Ο περαιτέρω ειδικότερος εργαστηριακός έλεγχος που διενεργήθηκε προς την πλευρά
του μεταβολικού νοσήματος με μέτρηση γαλακτικού και πυροσταφυλικού οξέος στον
ορό και το εγκεφαλονωτιαίο υγρό και με οργανικά οξέα ούρων, επίσης δεν κατέδειξε
παθολογικά ευρήματα, όπως επίσης και η εξέταση του εγκεφαλονωτιαίου υγρού.
Με βάση τη σοβαρότατη υποψία και τις σαφείς ενδείξεις που υπήρχαν από το περιφερειακό
νοσοκομείο από όπου παραπέμφθηκε το αγόρι -με βάση τα ευρήματα της μαγνητικής
τομογραφίας εγκεφάλου- διενεργήθηκε έλεγχος των VLCFA στο πλάσμα και τα αποτελέσματά
του ήταν συμβατά με Χ-ALD, όπως χαρακτηριστικά φαίνεται παρακάτω:
- C 22:0 56 μmοl/l (φτ: 41-119, φορείς Χ-ΑLD: 41-107, Χ-ΑLD: 32-76)
- C 24:0 90 μmοl/l (φτ: 33-84, φορείς Χ-ΑLD: 47-102, Χ-ALD: 44-115)
- C 26:0 3,98 μmol/l (φτ: 0,45-1,32, φορείς Χ-ΑLD: 1,11-2,93, Χ-ΑLD: 2-4,1)
- C 24:0/C 22:0 1,61 μmol/l (φτ: 0,57-0,92, φορείς Χ-ALD: 0,84-1,33, Χ-ALD:
1-1,8)
- C 26:0/ C22:0 0,071 μmol/l (φτ: <0,02, φορείς Χ-ΑLD: 0,02-0,04, Χ-ΑLD:
0.04-0,1)
Επίσης, διενεργήθηκε εκ νέου μαγνητική τομογραφία εγκεφάλου, η οποία έδειξε
χαρακτηριστικά συμμετρικές αλλοιώσεις με παθολογική ένταση, που αφορούν στην
οπίσθια περικοιλιακή λευκή ουσία και το σπληνίο του μεσολοβίου, όπως φαίνεται
στην εικόνα 1.
Τέλος, η εξέταση του ασθενούς συμπληρώθηκε με οφθαλμολογική εκτίμηση, εκτίμηση
της οπτικής οξύτητας, ΩΡΛ εκτίμηση, ακουολογικό έλεγχο και καρδιολογική εκτίμηση,
χωρίς να διαπιστωθούν πουθενά παθολογικά ευρήματα. Επίσης, διενεργήθηκε έλεγχος
νοημοσύνης (με τη μέθοδο WISC III), που κατέδειξε δείκτη νοημοσύνης στα επίπεδα
του 103.
Ο εργαστηριακός έλεγχος συμπληρώθηκε με ορμονολογικό έλεγχο των επινεφριδίων,
που κατέδειξε κορτιζόλη 10,9 μg/dl (φτ: 5,8-25) και ΑCΤΗ 131 pg/ml (φτ: 5,8-25).
Λόγω του τρόπου κληρονομικότητας της νόσου, προχωρήσαμε στον έλεγχο και των
άλλων μελών της οικογένειας του αγοριού. Ο κλινικός και εργαστηριακός έλεγχος
του μικρότερου αδελφού, ο οποίος ήταν φαινοτυπικά απόλυτα υγιής, κατέδειξε ευρήματα
συμβατά με Χ-ALD από τον έλεγχο των VLCFA στο πλάσμα και οριακά αυξημένη τιμή
ΑCΤΗ (ΑCΤΗ 55 pg/ml). Ο υπόλοιπος έλεγχος ήταν απολύτως φυσιολογικός, συμπεριλαμβανομένου
και της ΜRΙ εγκεφάλου και του δείκτη νοημοσύνης (δείκτης νοημοσύνης 102).
Ο κλινικός και εργαστηριακός έλεγχος του μεγαλύτερου ετεροθαλούς αδελφού ήταν
αρνητικός. Κατά τον κλινικοεργαστηριακό έλεγχο της μικρότερης αδελφής διαπιστώθηκαν
παθολογικές τιμές VLCFA στο πλάσμα σε επίπεδα συμβατά με ετερόζυγο Χ-ΑLD, γεγονός
το οποίο προέκυψε και μετά τον εργαστηριακό έλεγχο της μητέρας.
Κατά τον προγραμματισμένο επανέλεγχο του αγοριού και του μικρότερου πάσχοντος
αδελφού του, 5 μήνες μετά (το Φεβρουάριο του 2003), ενώ ο δεύτερος δεν παρουσίασε
διαφοροποίηση, ο ασθενής μας παρουσίασε κατά τη νευρολογική εκτίμηση ήπια πυραμιδική
συνδρομή στα κάτω άκρα και στον απεικονιστικό έλεγχο του εγκεφάλου με ΜRΙ, επιδείνωση
των ευρημάτων και κατάληψη και πρόσθιων περιοχών του εγκεφάλου (εικόνα 2).
Με βάση τη διεθνή βιβλιογραφία και τα μέχρι σήμερα δεδομένα στην αντιμετώπιση
της νόσου, αποφασίσθηκε τα δύο πάσχοντα αγόρια -ο ασθενής μας και ο μικρότερος
αδελφός του- να τεθούν σε δίαιτα με Lorentzo's oil, πράγμα το οποίο έγινε με
την πρώτη διάγνωσή τους και επιπρόσθετα στον ασθενή μας, μετά τη διαπίστωση
της επιδείνωσης της νόσου κατά τον επανέλεγχο, να προχωρήσουμε άμεσα στη μεταμόσχευση
μυελού.


ΣΥΖΗΤΗΣΗ
Η αδρενολευκοδυστροφία (Χ-ΑLD) μπορεί να παρουσιασθεί στους άρρενες με 6 διαφορετικούς
φαινοτύπους, από τους οποίους χαρακτηριστικότεροι και συχνότεροι είναι η παιδική
εγκεφαλική μορφή (childhood cerebral - CCER), η αδρενομυελονευροπάθεια (ΑΜΝ)
και η μορφή με νόσο Αddison μόνο (πίνακας 1). Αντίθετα, στα θήλεα μπορεί να
παρουσιασθεί με 5 διαφορετικούς φαινοτύπους, από τους οποίους χαρακτηριστικότεροι
είναι η ασυμπτωματική μορφή και η ήπια μυελοπάθεια (πίνακας 2).[6,7,12]
Τα αρχικά συμπτώματα με τα οποία μπορεί να προβάλει η εγκεφαλική μορφή της νόσου
(CCER) είναι συνήθως οι μαθησιακές δυσκολίες, οι διαταραχές συμπεριφοράς, η
βλάβη στην ακοή ή την όραση, και σπανιότερα η άνοια, οι σπασμοί, ο στραβισμός
και η διπλωπία, ή η δυσχέρεια βάδισης, ομιλίας ή γραφής. Σπανιότατα, η νόσος
μπορεί να προβάλει αρχικά με κεφαλαλγίες, καθ' έξιν κινήσεις (tics), ακράτεια
ούρων ή αυξημένη ενδοκράνια πίεση. Η κλινική εικόνα της Χ-ALD μπορεί να περιλαμβάνει
συμπτώματα από διάφορα συστήματα, αλλά κυρίως αφορά σε συμπτώματα από τα όργανα
"στόχους" της νόσου, δηλαδή νευρολογικές και αναπτυξιακές ανωμαλίες,
ενδοκρινολογικές διαταραχές ή διαταραχές στην ακοή και στην όραση (πίνακας 3).
Η πρόγνωση της Χ-ΑLD εξαρτάται κατά μεγάλο ποσοστό από την ηλικία έναρξης και
διάγνωσης της νόσου, σε συνδυασμό με τη βαρύτητα των ευρημάτων της ΜRΙ εγκεφάλου
τότε.[13] Έτσι, ασθενείς με πολλαπλά και εκτεταμένα ευρήματα, κατά 70-80% επιδεινώνονται
ανεξάρτητα από την ηλικία και μόνο 1 στους 10 παραμένουν νευρολογικά σταθεροί,
ενώ ασθενείς με μέτρια ευρήματα, κατά 60% επιδεινώνονται ανεξάρτητα από την
ηλικία και παρουσιάζουν μεγαλύτερη επιβίωση. Αντιθέτως, ασθενείς ηλικίας 3-7
ετών με ήπια ευρήματα, έχουν 30% πιθανότητα να αναπτύξουν εγκεφαλική συμμετοχή,
ενώ ίδιας κατηγορίας ασθενείς ηλικίας 7-10 ετών, μόλις 10% και μετά την ηλικία
των 10 ετών είναι πολύ σπάνιο να αναπτύξουν εγκεφαλική συμμετοχή.
Όσον αφορά την αντιμετώπιση της Χ-ΑLD, αυτή μπορεί να έχει κατ' αρχήν το χαρακτήρα
της πρόληψης, με την εξέταση όλων των μελών της οικογένειας του πάσχοντος, την
παροχή γενετικών συμβουλών και τον προγεννητικό έλεγχο, καθώς επίσης, ιδιαίτερα
σημαντική στην αντιμετώπιση της νόσου είναι η παρακολούθηση του ασθενούς, με
εκτίμηση και αξιολόγηση ανά εξάμηνο με ΜRΙ εγκεφάλου, με VLCFA στο πλάσμα και
ενδοκρινολογικό έλεγχο, με διενέργεια ελέγχου νοημοσύνης, νευρολογικής, ακουολογικής
και οφθαλμολογικής εκτίμησης και τέλος, με έλεγχο για πιθανές ανεπιθύμητες ενέργειες
από τη θεραπεία.[6,14]
Η θεραπεία της νόσου (πίνακας 4) αυτή καθ' αυτή, έγκειται στην αντιμετώπιση
της επινεφριδιακής ανεπάρκειας, της νευρολογικής προσβολής και στην υποστηρικτική
θεραπεία του ασθενούς και της οικογένειάς του (με ψυχολόγους, ιατρείο πόνου,
χειρουργούς, διαιτολόγους κ.ά.). Στο σημαντικό τομέα της νευρολογικής προσβολής,
έχουν χρησιμοποιηθεί διαιτητικοί παράγοντες και παρεμβάσεις χωρίς ιδιαίτερα
αποτελέσματα, εκτός από το Lorentzo's oil. Η πλασμαφαίρεση που έχει δοκιμασθεί
δεν έχει αποδώσει, η ανοσοκαταστολή, καθώς και η γονιδιακή θεραπεία βρίσκονται
υπό έρευνα, ενώ η θεραπεία της νόσου φαίνεται ότι επιτυγχάνεται μόνο με τη μεταμόσχευση
του μυελού των οστών, με φτωχή όμως μέχρι σήμερα εμπειρία, πρόγνωση και ποσοστά
επιτυχίας.
Το Lorentzo's oil αποτελεί μείγμα 4:1 γλυκερυλ-τριοελαϊκό έλαιο και γλυκερυλ-τριερουκικό
έλαιο, και ο τρόπος δράσης του βασίζεται στο ερουκικό οξύ (ω-9-μονοακόρεστο
22C-λιπαρό οξύ), που ανταγωνίζεται το ρυθμό ενδογενούς σύνθεσης VLCFA στο ενζυμικό
σύστημα των μικροσωματίων.[15,16] Η χορήγησή του ενδείκνυται σε ασθενείς χωρίς
νευρολογική συμπτωματολογία ή συμμετοχή (ΜRΙ εγκεφάλου: κ.φ.), στους οποίους,
σύμφωνα με μελέτες, μειώνει τη συχνότητα και τη βαρύτητα της νευρολογικής διαταραχής
(χωρίς όμως να βελτιώνει την ενδοκρινική λειτουργία). Πρέπει, επίσης, χαρακτηριστικά
να σημειωθεί ότι το Lorentzo's oil εντός 4 μηνών από την έναρξη της χορήγησής
του, επαναφέρει τα επίπεδα των VLCFA του πλάσματος σε φυσιολογικά επίπεδα, ενώ
δεν περνά τον αιματοεγκεφαλτκό φραγμό και έτσι δεν έχει καμία επίδραση στα VLCFA
του εγκεφάλου.
Η μεταμόσχευση του μυελού των οστών αποτελεί την περισσότερο υποσχόμενη και
αποτελεσματική μορφή θεραπείας της Χ-ΑLD.[18,19,20,21] Μέχρι σήμερα έχουν διενεργηθεί
πάνω από 200 μεταμοσχεύσεις του μυελού των οστών για τη θεραπεία ασθενών με
Χ-ΑLD, με θνητότητα ιδιαίτερα υψηλή που φθάνει το 40%, αλλά μπορεί να επιτύχει
τη σταθεροποίηση (σε ποσοστό έως και 40%) ή και την αναστροφή της νόσου (σε
ποσοστό έως και 20%). Ο ακριβής μηχανισμός με τον οποίο τα μεταμοσχευμένα κύτταρα
δρουν, παραμένει σε πολλά σημεία άγνωστος φαίνεται, όμως, ότι διέρχονται στον
εγκέφαλο και παράγουν μικρογλοιακά κύτταρα και νευρικό ιστό, με αποτέλεσμα να
εκφράζουν το παθολογικό γονίδιο και ως εκ τούτου, να γίνεται ο μεταβολισμός
και συνεπώς η ελάττωση της στάθμης των VLCFA στον οργανισμό. Τέλος, φαίνεται
ότι μειώνουν τη φλεγμονώδη αντίδραση στον εγκέφαλο. Ενδείκνυται για τη θεραπεία
ασθενών οι οποίοι βρίσκονται σε πρώιμα στάδια εγκεφαλικής προσβολής και συμμετοχής
με βάση τα ευρήματα από την ΜRΙ εγκεφάλου και τις νευροψυχιατρικές εξετάσεις
(ΙQ>80), με όσο το δυνατόν συμβατό για ΗLΑ δότη. Αντενδείκνυται σε ασθενείς
με ταχέως εξελισσόμενη ή βαριά εγκεφαλική συμμετοχή και σε ασυμπτωματικούς ασθενείς
με φυσιολογική ΜRΙ εγκεφάλου, ενώ δεν έχει εφαρμοσθεί σε ασθενείς ασυμπτωματικούς
με παθολογική ΜRΙ εγκεφάλου και σε ασθενείς με ΑΜΝ, λόγω του υψηλού ποσοστού
θνητότητας σε σχέση με την καλή εξέλιξη των τελευταίων.
Η Χ-ΑLD πρέπει να τίθεται στη διαφοροδιαγνωστική σκέψη του γενικού παιδίατρου
σε περιπτώσεις αγοριών που παρουσιάζουν προοδευτική διαταραχή στη συμπεριφορά,
έκπτωση της μαθησιακής ικανότητας, νευρολογικές διαταραχές που αρχίζουν στην
ηλικία των 3 ετών, άρρενες με νόσο Αddison που δεν έχει καθορισθεί το αίτιο,
ασθενείς με προοδευτική σπαστική παραπάρεση (ΑΜΝ - ΜS), ασθενείς που η επινεφριδιακή
ανεπάρκεια συνδυάζεται με νευρολογική διαταραχή ή έκπτωση και τέλος, φυσικά,
άτομα υψηλού κινδύνου λόγω συγγένειας με διαγνωσθέντες ασθενείς.

Ευχαριστούμε για τη συμβολή τους στη διαγνωστική προσπέλαση του περιστατικού την κα Μιχελακάκη στο Ινστιτούτο Υγείας του Παιδιού και τον κο Νίκα στο τμήμα του μαγνητικού τομογράφου.
Adrenoleucodystrophy
(ALD): A case report
A. Papaloukas, D. Lazopoulou, S. Youroucos, E. Drogari, G. Chrousos
(Ann Clin Paediatr Univ Atheniensis 2004, 51(1):58-65)
Adrenoleucodystrophy (ALD) is a genetically determined disorder of the function of the peroxisomes and is inherited by X-linked recessive transmission. A case of ALD in a 9,5 year old boy, who was sent from another hospital and admitted in our clinic for investigation of a seizures episode, is described. Diagnosis was suspected by the symptoms and the cerebral Magnetic Resonance Imaging (MRI), where characteristic findings to the disease were identified. Diagnosis was established by the finding of high levels of Very Long Chain Fatty Acids (VLCFA) in serum in correlation to the findings of the cerebral MRI.
ΒΙΒΛΙΟΓΡΑΦΙΑ
1. HugoW Moser, Kirby D Smith, Paul A Watkins et al. X-linked Adrenoleucodystrophy.
Metabolic and Inheritance Diseases, 131:3257-3293.
2. Hugo W Moser. Adrenoleucodystrophy (X-linked). Nelson Textbook of Pediatrics
2000, 72.2:367-370.
3. Maja Di Rocco, Laura Doria-Lamba, Ubaldo Caruso. Monogygotic Twins with X-linked
Adrenoleucodystrophy and Different Phenotypes. Ann Neurol 2001 50.3:424.
4. Sobue G, Ueno-Natsukari I, Okamoto H et al. Phenotypic heterogeneity of an
adult form of adrenoleucodystrophy in monogygotic twins. Ann Neurol 1991; 36:912-915.
5. Korenke GC, Fucks S, Krasemann E et al. Cerebral adrenoleucodystrophy (ADL)
in only one of monozygous twins with an identical ADL genotype. Ann Neurol 1996;
40:254-257.
6. Hugo W Moser. Adrenoleucodystrophy: phenotype, genetics, pathogenesis and
therapy. Brain 1997; 120:1485-1508.
7. Moser HW, Bergin A, Naidu S et al. Adrenoleucodystrophy. Endocrinol Metab
Clin North Am 1991; 20:297-317.
8. Moser HW, Moser AB, Kawamura N et al. Adrenoleucodystrophy: elevated C26
fatty acid in cultured skin fibroblasts. Ann Neurol 1980; 7:542-549.
9. Moser HW, Moser AB, Prayer KK et al. Adrenoleucodystrophy: increased plasma
content of saturated very long chain fatty acids. Neurology 1981; 31:1241-1246.
10. Wanders RGA, van Roermund CWT, Lageweg W et al. X-linked Adrenoleucodystrophy:
Biochemical diagnosis and enzyme defect. J Inherit Metab Dis 1992; 15:634-644.
11. Moser AB, Kreiter N, Bezman L et al. Plasma Very Long Chain Fatty Acids
in 3000 Peroxisome Disease Patients and 29000 Controls. Ann Neurol 1999; 45:100-110.
12. Korenke GC, Roth C, Krasemann E et al. Variability of endocrine logical
difunction in 55 patients with X-linked adrenoleucodystrophy: clinical, laboratory
and genetic findings. Eur J Endocr 1997; 137:40-47.
13. Moser HW, Loes DJ, Melhem ER et al. X-linked Adrenoleucodystrophy: Overview
and Prognosis as a Function of Age and Brain Magnetic Resonance Imaging Abnormality.
A Study Involving 372 Patients. Neuropediatrics 2000; 31:227-239.
14. Brown FR III, van DuynMA, Moser AB et al. Adrenoleucodystrophy: effects
of a dietary restriction and of administration of carnitine and clofibrate on
clinical status and plasma Fatty acids. Johns Hopkins Med J 1982; 151:164-172.
15. Moser HW. Treatment of X-linked adrenoleucodystrophy with Lorentzo's oil.
J Neurol Neurosurg Psychiatry 1999; 67:279-280.
16. Van Geel BM, Assies J, Havercort EB et al. Progression of abnormalities
in adrenomyelopathy and neurologically asymptomatic X-linked adrenoleucodystrophy
despite treatment with "Lorentzo's oil". J Neurol Neurosurg Psychiatry
1999; 67:290-299.
17. Inderjit Singh, Mushfiquddin Khan, Lyndon Key, et al. Lovastatin for X-linked
Adrenoleucodystrophy. New Engl J Med 1998; 339:702-703.
18. Shapiro E, Krivit W, Lockman L et al. Long term effect of bone-marrow transplantation
for childhood onset cerebral X-linked adrenoleucodystrophy. Lancet 2000; 356:713-718.
19. Peters C, Steward CG. Hematopoetic cell transplantation for inherited metabolic
diseases: an overview of outcomes and practice guidelines. Bone Mar Transpl
2003; 31:229-239.
20. Thomas Graf. Differentiation plasticity of hematopoetic cells. Blood 2002;
99:3089-3101.
21. Steindler DA, Pincus DW. Stem cells and neuropoiesis in the adult human
brain. Lancet 2002; 359:1047-1054.
