
Μυελοδυσπλαστικά και
μυελοϋπερπλαστικά σύνδρομα
της παιδικής ηλικίας
Μ. Μοσχόβη[1]
Γ. Τρίμης[1]
B. Τουλιάτου[2]
K. Στεφανάκη[3]
Β. Κίτρα[4]
Φ. Τζωρτζάτου-Σταθοπούλου[1]
[1]
Μονάδα Αιματολογίας - Ογκολογίας, Α΄ Παιδιατρικής Κλινικής Πανεπιστημίου Αθηνών
Νοσοκομείου Παίδων "Αγία Σοφία"
[2] Τμήμα Γενετικής Πανεπιστημίου Αθηνών
[3] Παθολογοανατομικό Εργαστήριο Νοσοκομείου Παίδων "Αγία Σοφία"
[4] Μονάδα Μεταμόσχευσης Αιμοποιητικών Κυττάρων Νοσοκομείου Παίδων "Αγία
Σοφία"
Υποβλήθηκε:
9/11/2003
ΠΕΡΙΛΗΨΗ
Τα μυελοδυσπλαστικά (ΜΔΣ) και τα μυελοϋπερπλαστικά (ΜΥΣ) σύνδρομα περιλαμβάνουν
ένα ετερογενές σύνολο κλωνικών διαταραχών του αρχέγονου αιμοποιητικού κυττάρου,
που λόγω της σπανιότητάς τους δεν έχουν μελετηθεί επαρκώς στην παιδική ηλικία.
Στην παρούσα μελέτη γίνεται καταγραφή των ιδιαίτερων χαρακτηριστικών των ΜΔΣ
και ΜΥΣ που διαγνώστηκαν και αντιμετωπίστηκαν στη Μονάδα μας την τελευταία πενταετία
(1998-2002). Παράλληλα γίνεται ανασκόπηση της βιβλιογραφίας.
Υλικό της μελέτης αποτέλεσαν 8 παιδιά (6 αγόρια-2 κορίτσια) ηλικίας 2-18 χρονών
(μέση ηλικία 9 ετών) με ΜΔΣ (6 πρωτοπαθή-2 δευτεροπαθή) και 5 παιδιά (3 αγόρια-2
κορίτσια) ηλικίας 1-13 ετών (μέση ηλικία 8 ετών) με ΜΥΣ. Παθολογικός κυτταρογενετικός
έλεγχος ανέκυψε σε 3/8 παιδιά με ΜΔΣ, ενώ τα 5/8 υποβλήθηκαν σε μεταμόσχευση
μυελού των οστών (ΜΜΟ). Να σημειωθεί ότι 2/6 παιδιά με πρωτοπαθές ΜΔΣ εμφάνισαν
οξεία μυελοβλαστική λευχαιμία (ΟΜΛ) σε διάστημα 6 και 12 μηνών. Ο θάνατος επήλθε
σε 3/8 παιδιά, τα υπόλοιπα παραμένουν σε ύφεση για διάστημα 10 μήνες έως 4 χρόνια
(μέσο διάστημα 26 μήνες). Στους ασθενείς με ΜΥΣ χορηγήθηκε υδροξυουρία σε 2/5
παιδιά, ενώ το ένα παιδί με μυελομονοκυτταρική λαυχαιμία νεανικού τύπου (JMML)
υποβλήθηκε σε ΜΜΟ από ιστοσυμβατό συγγενή δότη 2 φορές, καθώς ενδιάμεσα ανέπτυξε
οξεία λεμφοβλαστική λευχαιμία (ΟΛΛ). Στα παιδιά με ιδιοπαθή θρομβοκυτταραιμία
(ΕΤ) χορηγήθηκε αναγκρελίδη. Όλοι οι ασθενείς είναι σε ύφεση 2.5-4 χρόνια (μέσο
διάστημα 38 μήνες).
Από τα ευρήματα της μελέτης φαίνεται ότι τα ΜΔΣ στα παιδιά εκδηλώνονται πιο
συχνά με τις επιθετικές μορφές, συνδυάζονται με κυτταρογενετικές ανωμαλίες,
συχνά μεταπίπτουν σε ΟΜΛ, ενώ θεραπεία εκλογής είναι η αλλογενής ΜΜΟ. Τα αποτελέσματα
της ΜΜΟ είναι αρκετά ευνοϊκά στα πρωτοπαθή, ενώ είναι απογοητευτικά στα δευτεροπαθή
ΜΔΣ. Τα ΜΥΣ της παιδικής ηλικίας έχουν συνήθως σχετικά καλή πορεία και σπανιότερα
μεταπίπτουν σε λευχαιμία. Σταθερό κλινικό σημείο είναι η σπληνομεγαλία. Πλην
της χρόνιας μυελογενούς λευχαιμίας τύπου ενηλίκου (CML), στα ΜΥΣ εντοπίζονται
λιγότερο συχνά γενετικές διαταραχές. Η αλλογενής ΜΜΟ αποτελεί τη θεραπεία εκλογής
στη JMML και τη CML, ενώ η αναγκρελίδη καθιερώνεται ως φάρμακο εκλογής στην
Ιδιοπαθή θρομβοκυτταραιμία (ΕΤ) χωρίς παρενέργειες. (Δελτ Α΄ Παιδιατρ Κλιν Πανεπ
Αθηνών 2004, 51(1):39-51
Λέξεις ευρετηριασμού: μυελοδυσπλαστικά σύνδρομα, μυελοϋπερπλαστικά σύνδρομα, παιδική ηλικία, λευχαιμία.
ΕΙΣΑΓΩΓΗ
Τα μυελοδυσπλαστικά (ΜΔΣ) και τα μυελοϋπερπλαστικά (ΜΥΣ) σύνδρομα αποτελούν
σπάνια οντότητα για την παιδική ηλικία. Είναι ένα ετερογενές σύνολο κλωνικών
διαταραχών του αρχέγονου αιμοποιητικού κυττάρου οι οποίες συγκαταλέγονται στις
προλευχαιμικές καταστάσεις.[1] Τα ΜΔΣ χαρακτηρίζονται από κυτταροπενία μίας
ή περισσοτέρων σειρών διάρκειας >6 μηνών που συνήθως συνδυάζονται με υπερκυτταρικό
μυελό (μη αποτελεσματική αιμοποίηση) και αυξημένο κίνδυνο να μεταπέσουν σε οξεία
μυελοβλαστική λευχαιμία (ΟΜΛ). Τα ΜΥΣ χαρακτηρίζονται από υπέρμετρη ανάπτυξη
μίας ή περισσοτέρων σειρών στο μυελό των οστών και στο περιφερικό αίμα, που
σχεδόν πάντοτε συνοδεύεται από σπληνομεγαλία. Περιορισμένα επιδημιολογικά στοιχεία
υπάρχουν από μικρές σειρές ασθενών με ΜΔΣ, όπου υπολογίζεται ότι αυτά αντιπροσωπεύουν
το 3%-9% των παιδιατρικών αιματολογικών κακοηθειών και με ετήσια επίπτωση από
1 έως 4 νέα περιστατικά ανά 1.000.000 παιδιά <15 χρονών. Ενώ, δεν υπάρχουν
επιδημιολογικές αναφορές για ΜΥΣ σε παιδιά. Ως προδιαθεσικοί παράγοντες ανάπτυξης
ΜΔΣ ή ΜΥΣ έχουν αναγνωριστεί σύνδρομα που συνδυάζονται με μυελική ανεπάρκεια
(πχ. Down, Kostmann, Fanconi, Schwachmann), ανοσοανεπάρκειες, χρωμοσωμικές ανωμαλίες
(πχ. τρισωμία 8, μονοσωμία 7), χορηγηθείσα χημειοθεραπεία ή ακτινοθεραπεία για
άλλες κακοήθειες.[2]
Εκτός από τη σπανιότητα αυτών των νοσημάτων στα παιδιά, μεγάλη δυσκολία στη
μελέτη τους και στον προσδιορισμό της πραγματικής τους συχνότητας δημιουργεί
η απουσία ομοφωνίας όσον αφορά στην κατάταξή τους. Μέχρι σήμερα έχουν προταθεί
4 συστήματα κατάταξης των συνδρόμων αυτών, η κατά FAB (French-American-British),
η κατά WHO (World Health Organization), η κατά CCC (Category-Cytology-Cytogenetics)
και η κατά IPSS (International Prognostic Scoring System). Οι διαφοροποιήσεις
μεταξύ των τεσσάρων αυτών συστημάτων είναι στην πραγματικότητα μικρές. Φαίνεται
ότι η κατά FAB κατάταξη, μετά τη μερική αναθεώρηση των κριτηρίων της το 1997,
είναι αυτή που ξεχωρίζει καλύτερα τις διάφορες μορφές ΜΔΣ και ΜΥΣ της παιδικής
ηλικίας.[3] Τα νοσήματα αυτά συνιστούν ένα σχετικά νέο πεδίο επιστημονικής έρευνας,
αφού μόλις τα τελευταία χρόνια αρχίζει να γίνεται κατανοητή η παθογένεια τους,
γι'αυτό το ενδιαφέρον των αιματολόγων έχει επικεντρωθεί στους ενήλικες στους
οποίους είναι πολύ πιο συχνά. Οι διαταραχές αυτές στα παιδιά παρουσιάζουν ιδιαιτερότητες
που τις καθιστούν ξέχωρες παθολογικές οντότητες, με συνέπεια να μη μπορεί να
εφαρμοστεί άκριτα η διαγνωστική και θεραπευτική προσέγγιση που χρησιμοποιείται
για τα ΜΔΣ και ΜΥΣ των ενηλίκων. [1,4]
Οι παραπάνω λόγοι και το γεγονός ότι δεν υπάρχουν πολλές καταγεγραμμένες σειρές
ασθενών της παιδικής ηλικίας αποτέλεσε ερέθισμα ώστε να γίνει καταγραφή και
μελέτη των ιδιαίτερων χαρακτηριστικών των ΜΔΣ και ΜΥΣ που διαγνώστηκαν και αντιμετωπίστηκαν
στη Μονάδα μας την τελευταία πενταετία (1998-2002). Στόχος είναι η ευαισθητοποίηση
των παιδιάτρων στην ύπαρξη αυτών των συνδρόμων και η έγκαιρη ανίχνευση τους
ώστε με την κατάλληλη παρακολούθηση να προληφθεί η εξέλιξή τους σε επιθετικότερες
μορφές, στις οποίες συμπεριλαμβάνεται η οξεία λευχαιμία.
ΥΛΙΚΟ
ΚΑΙ ΜΕΘΟΔΟΙ
Υλικό της παρούσης μελέτης αποτέλεσαν τα παιδιά με ΜΔΣ ή ΜΥΣ τα οποία νοσηλεύτηκαν
στην Μονάδα Αιματολογίας-Ογκολογίας της A' Παιδιατρικής Κλινικής του Πανεπιστημίου
Αθηνών την τελευταία πενταετία (1998-2002). Έγινε καταγραφή των επιδημιολογικών
στοιχείων, της κλινικής προβολής, των εργαστηριακών ευρημάτων, της θεραπείας
και της έκβασης του νοσήματος σε κάθε ασθενή. Τα ΜΔΣ διαχωρίστηκαν: α) σε πρωτοπαθή,
όταν οι ασθενείς δεν έπασχαν από άλλο προϋπάρχον νόσημα, και β) σε δευτεροπαθή,
όταν οι ασθενείς είχαν υποβληθεί κατά το παρελθόν σε θεραπεία για συμπαγή όγκο
ή οξεία λευχαιμία. Η κατάταξη των περιστατικών έγινε με τα κριτήρια κατά FAB
που, παρά τις όποιες αμφισβητήσεις που έχουν δεχθεί, παραμένουν τα επικρατέστερα
(Πίνακας 1).
ΑΠΟΤΕΛΕΣΜΑΤΑ
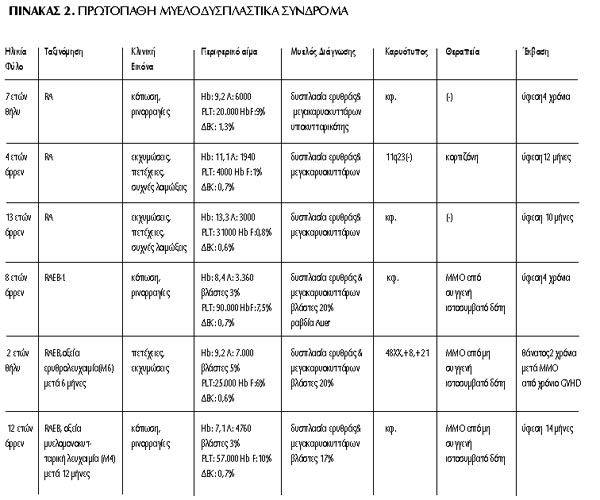
Συνολικά νοσηλεύτηκαν 8 παιδιά (6 αγόρια-2 κορίτσια) ηλικίας 2-18
χρονών (μέση ηλικία 9 ετών) με ΜΔΣ (6 πρωτοπαθή-2 δευτεροπαθή) και 5 παιδιά
(3 αγόρια-2 κορίτσια) ηλικίας 1-13 ετών (μέση ηλικία 8 ετών) με ΜΥΣ. Από την
ομάδα των ΜΔΣ, σε 3 παιδιά τέθηκε η διάγνωση της πρωτοπαθούς ανθεκτικής αναιμίας
(RΑ), σε 2 παιδιά της πρωτοπαθούς ανθεκτικής αναιμίας με περίσσεια βλαστών (RAEB),
σε 1 παιδί της πρωτοπαθούς ανθεκτικής αναιμίας με περίσσεια βλαστών σε μετατροπή
(RAEB-t) και σε 2 παιδιά της δευτεροπαθούς RAEB-t. Τα δύο τελευταία παιδιά είχαν
εκδηλώσει οξεία λεμφοβλαστική λευχαιμία από Β-κύτταρο προ 6ετίας και λέμφωμα
Hodgkin προ 5ετίας αντίστοιχα και είχαν υποβληθεί σε χημειοθεραπεία χωρίς ακτινοθεραπεία.
Από την ομάδα των ΜΥΣ, σε 2 παιδιά διαγνώστηκε μυελομονοκυτταρική λευχαιμία
νεανικού τύπου (JMML), σε 1 παιδί χρόνια μυελογενής λευχαιμία τύπου ενηλίκου
(CML) και σε 2 παιδιά ιδιοπαθής θρομβοκυτταραιμία (ΕΤ).
Η κλινική εικόνα των ασθενών με ΜΔΣ κατά τη διάγνωση ήταν αποτέλεσμα της κυτταροπενίας
μίας ή περισσοτέρων σειρών του περιφερικού αίματος (εύκολη κόπωση, ωχρότητα,
συχνές λοιμώξεις, πετέχειες, εκχυμώσεις, ρινορραγίες). Στο μυελόγραμμα διαπιστώθηκε
δυσπλασία της ερυθράς σειράς και δυσπλασία των μεγακαρυοκυττάρων σε όλους τους
ασθενείς και παρουσία βλαστών 17-25% σε 5/8 ασθενείς. Από το περιφερικό αίμα
διαπιστώθηκε θρομβοπενία σε όλα τα παιδιά, αναιμία με χαμηλά ΔΕΚ και παρουσία
βλαστών 3-5% σε 5/8 παιδιά, υψηλή τιμή HbF (6-10%) σε 4/8 παιδιά και λευκοπενία
σε ένα παιδί. Από τον κυτταρογενετικό έλεγχο σε ένα παιδί με πρωτοπαθή RA ανευρέθηκε
έλλειμμα στην περιοχή 11q23, σε ένα με πρωτοπαθή RAEB ανευρέθηκε τρισωμία 8
και τρισωμία 21 και σε ένα παιδί με δευτεροπαθές RAEB-t ανευρέθηκε μετάθεση
(5;17), μονοσωμία 7 και τρισωμία 8. Όλα τα παιδιά, πλην αυτών με πρωτοπαθή RA,
υποβλήθηκαν σε μεταμόσχευση μυελού των οστών (ΜΜΟ) από απόλυτα συμβατό συγγενή
ή μη συγγενή δότη. Ένα παιδί με δευτεροπαθές ΜΔΣ υποβλήθηκε σε θεραπεία με κυκλοσπορίνη
και αντιθυμοκυτταρική σφαιρίνη χωρίς ικανοποιητική ανταπόκριση και ακολούθως,
επειδή δεν υπήρχε συμβατός συγγενής ή μη συγγενής δότης, υποβλήθηκε σε ΜΜΟ από
απλοταυτόσημο συγγενή δότη. Να σημειωθεί ότι σε 2/6 παιδιά το πρωτοπαθές ΜΔΣ
σε διάστημα 6 και 12 μηνών μετέπεσε σε οξεία μυελοβλαστική λευχαιμία (ΟΜΛ) τύπου
ερυθρολευχαιμίας (Μ6) και μυελομονοκυτταρικής (Μ4) αντίστοιχα, καθ' όσον χρόνο
αναζητείτο συμβατός δότης για ΜΜΟ.
Η έκβαση των παιδιών με πρωτοπαθές ΜΔΣ ήταν καλύτερη από αυτήν των παιδιών με
δευτεροπαθές ΜΔΣ. Τα 2/3 παιδιά με πρωτοπαθή RA μετέπεσαν σε ύφεση χωρίς να
λάβουν κάποια αγωγή. Αξιοσημείωτη είναι η περίπτωση ασθενούς με πρωτοπαθή RAEB,
που ενώ παρουσίασε αυτόματη ύφεση με βλάστες <1% στο μυελό και είχε φυσιολογικό
καρυότυπο, 6 μήνες αργότερα παρουσίασε οξεία ερυθρολευχαιμία (Μ6), υποβλήθηκε
σε αλλογενή ΜΜΟ και κατέληξε 2 χρόνια μετά τη μεταμόσχευση από χρόνιο GVHD.
Συνοπτικά τα στοιχεία όλων των ασθενών με ΜΔΣ καταγράφονται στους Πίνακες 2
και 3.
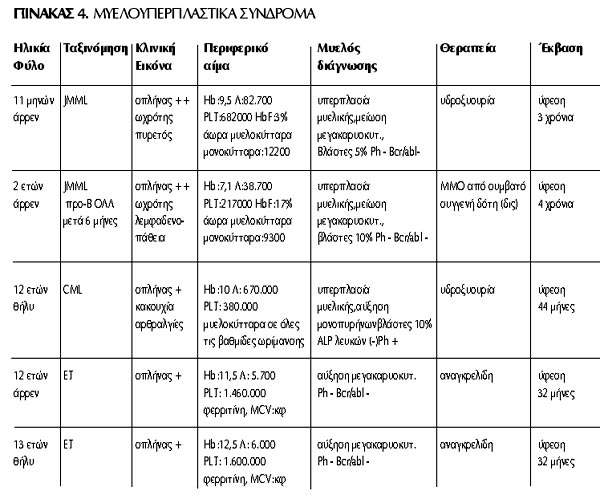
Η κλινική εικόνα κατά τη διάγνωση των παιδιών με ΜΥΣ ήταν πιο ήπια απ'ότι στα
ΜΔΣ. Σταθερό εύρημα ήταν η σπληνομεγαλία. Στα 2 παιδιά με JMML συνυπήρχε ωχρότητα,
το ένα παρουσίαζε πυρετό και το άλλο γενικευμένη λεμφαδενοπάθεια. Ο ασθενής
με τη CML εμφάνισε κακουχία και αρθραλγίες, ενώ τα 2 παιδιά με την ΕΤ ήταν ασυμπτωματικά
και η διάγνωση τέθηκε στα πλαίσια διερεύνησης της σπληνομεγαλίας. Στο περιφερικό
αίμα των ασθενών με JMML διαπιστώθηκε αναιμία, αυξημένα λευκά και μονοκύτταρα,
άωρα μυελοκύτταρα, ενώ το ένα παιδί είχε και αυξημένη HbF (17%). Τα ευρήματα
του περιφερικού αίματος στον ασθενή με τη CML ήταν λευκοκυττάρωση, ιδιαίτερα
εκσεσημασμένη κατά τη διάρκεια της βλαστικής κρίσης (670.000/μl), και παρουσία
μυελοκυττάρων σε όλες τις βαθμίδες ωρίμανσης, ενώ στην ΕΤ το μόνο εύρημα ήταν
η θρομβοκυττάρωση. Ο μέσος όγκος ερυθρών (MCV) και η φερριτίνη ορού ήταν φυσιολογικά
και στους 2 ασθενείς με ΕΤ. Στο μυελόγραμμα των ασθενών με JMML διαπιστώθηκε
υπερπλασία της μυελικής και μείωση της μεγακαρυοκυτταρικής σειράς, βλάστες 5-10%,
ενώ στην κυτταρογενετική μελέτη του μυελού διαπιστώθηκε απουσία του χρωμοσώματος
Philadelphia (Ph) και της σύντηξης των γόνων bcr/abl. Στο μυελόγραμμα του παιδιού
με τη CML διαπιστώθηκε υπερπλασία της μυελικής σειράς με αύξηση των μονοπυρήνων,
βλάστες 10%, ελαττωμένη δραστηριότητα της αλκαλικής φωσφατάσης των λευκών, ενώ
στην κυτταρογενετική μελέτη του μυελού εντοπίστηκε το χρωμόσωμα Ph. Τέλος, στο
μυελόγραμμα των ασθενών με ΕΤ διαπιστώθηκε υπερπλασία της μεγακαρυοκυτταρικής
σειράς, ενώ δεν ανιχνεύτηκε το χρωμόσωμα Ph. Ως θεραπεία χορηγήθηκε υδροξυουρία
στον ένα ασθενή με JMML και στο παιδί με τη CML, λόγω έλλειψης δότη για ΜΜΟ,
ενώ το άλλο παιδί με JMML υποβλήθηκε σε ΜΜΟ από ιστοσυμβατό συγγενή δότη. Στα
παιδιά με ΕΤ χορηγήθηκε αναγκρελίδη. Ενδιαφέρον είναι ότι το παιδί με JMML που
υποβλήθηκε σε ΜΜΟ 6 μήνες αργότερα ανέπτυξε οξεία λεμφοβλαστική λευχαιμία από
προ-Β κύτταρο, ακολούθως υποβλήθηκε σε δεύτερη μεταμόσχευση από τον ίδιο δότη
και έκτοτε παραμένει σε ύφεση επί 4 χρόνια. Η έκβαση των 5 παιδιών με ΜΥΣ είναι
ικανοποιητική, καθώς η νόσος είναι σε ύφεση για διάστημα 2.5-4 χρόνια σε όλους
τους ασθενείς (μέσο διάστημα 38 μήνες). Συνοπτικά τα στοιχεία όλων των ασθενών
με ΜΥΣ καταγράφονται στον πίνακα 4.
ΣΥΖΗΤΗΣΗ
Τα ΜΔΣ και τα ΜΥΣ στα παιδιά συγκαταλέγονται στις προλευχαιμικές καταστάσεις
εμφανίζοντας αυξημένη τάση εξέλιξης σε πλέον επιθετικά κλωνικού τύπου νοσήματα,
αν και μερικές φορές μπορεί να παρουσιάσουν και αυτόματη ύφεση. Η παθογένεια
των συνδρόμων παραμένει αδιευκρίνιστη. Οι παράγοντες οι οποίοι με τη δράση τους
δημιουργούν ή επιλέγουν υπέρ ενός παθολογικού κυτταρικού κλώνου, ο οποίος στη
συνέχεια αναπτύσσεται υπέρμετρα, είναι σε μεγάλο βαθμό άγνωστοι, όπως άγνωστο
παραμένει τι οδηγεί στην τελική επικράτηση συγκεκριμένων κλινικών και εργαστηριακών
ευρημάτων ανάλογα με την υποκατηγορία του ΜΔΣ ή ΜΥΣ.[1,5]

Παθογένεια ΜΔΣ και ΜΥΣ
Η πλέον σύγχρονη άποψη που φαίνεται να επικρατεί ως προς την παθογένεια
αυτών των συνδρόμων αναφέρεται στην πιθανή δράση κάποιου μεταλλαξιογόνου παράγοντα
στο πολυδύναμο αρχέγονο αιμοποιητικό κύτταρο της τροφοδοτικής αιμοποιητικής
δεξαμενής του μυελού των οστών (stem cell).[6] Ο παράγοντας αυτός μπορεί να
είναι άγνωστος, ή να αφορά σε προηγηθείσα χημειοθεραπεία, ακτινοθεραπεία και
κυτταρογενετική ανωμαλία που είναι δυνατόν να προδιαθέτει σε μυελική ανεπάρκεια
(πχ σύνδρομα ανοσοανεπάρκειας, νευροϊνωμάτωση τύπου Ι, Fanconi, Noonan, Down,
Bloom, Schwachmann, Kostmann). Το μεταλλαγμένο αυτό κύτταρο πλεονεκτεί των φυσιολογικών
όσον αφορά στην πολλαπλασιαστική του δυνατότητα έναντι των υπολοίπων φυσιολογικών
κυττάρων. Υπό την επίδραση εξωγενών ή άλλων παραγόντων του μικροπεριβάλλοντος
του μυελού των οστών, είναι δυνατό να εμφανιστούν ως επιφαινόμενα και άλλες
βλάβες στο γενετικό υλικό του παθολογικού κλώνου, με αποτέλεσμα την εξέλιξη
του υπάρχοντος νοσήματος σε επιθετικότερο τύπο ή την εκτροπή του σε οξεία λευχαιμία.
Η παθολογική αυτή διεργασία έχει ανοσολογική βάση και βρίσκεται κάτω από την
άμεση επίδραση των κυτταροκινών και κυρίως του παράγοντα νέκρωσης του όγκου
(TNF-α) και της ιντερλευκίνης-Ιβ (IL-Ιβ).[7] Η σύνθετη και αδιευκρίνιστη αυτή
δράση των κυτταροκινών επάγει τον πολλαπλασιασμό των CD34+, την αναστροφή του
λόγου CD4+/CD8+ και την αδρανοποίηση των ΝΚ κυττάρων. Επίσης στα ΜΔΣ παρατηρείται
αυξημένος αποπτωτικός θάνατος των πιο ώριμων θυγατρικών κυττάρων, ενώ στα ΜΥΣ
αναστολή της αποπτωτικής διαδικασίας.[8] Η αυξημένη απόπτωση των πρόδρομων κυττάρων
του μυελού των οστών στα ΜΔΣ θεωρείται ότι οφείλεται σε διαταραχή του υποδοχέα
κυτταρικού θανάτου (Fas) και του συνδετικού του μορίου (Fas ligand). Επιπρόσθετο
ρόλο φαίνεται να διαδραματίζουν κάποια ογκογονίδια και ογκοκατασταλτικά γονίδια
(ras, p53).[9]
Ο κεντρικός ρόλος, όμως, στο φαινόμενο της απόπτωσης αποδίδεται στα μιτοχόνδρια.
Βλάβες των μιτοχονδρίων, λόγω σημειακών μεταλλάξεων του μιτοχονδριακού DNA,
προκαλούν απελευθέρωση αποπτωτικών παραγόντων στο κυτταρόπλασμα, όπως το κυτόχρωμα
c και οι ελεύθερες ρίζες οξυγόνου και καταστροφή του συστήματος μεταφοράς ηλεκτρονίων
με τελικό αποτέλεσμα τη διακοπή παραγωγής ATP. Οι διεργασίες αυτές συντελούν
στην ενεργοποίηση των κασπασών, σύνολο πρωτεασών της κυστεϊνης, στη γένεση οξειδωτικού
stress στα πρόδρομα μυελικά κύτταρα και συνακόλουθα στον κυτταρικό θάνατό τους
ή πολύ σπανιότερα στη δημιουργία δακτυλιοειδών σιδηροβλαστών. Πειραματικά δεδομένα
όπως η μειωμένη κατανάλωση οξυγόνου από τα μονοπύρηνα του μυελού και η αυξημένη
συγκέντρωση ομοκυστεϊνης στον ορό ασθενών πασχόντων από ΜΔΣ είναι έμμεσοι δείκτες
της παραπάνω διαταραχής.[10] Υπάρχουν ενδείξεις ότι η διεργασία αυτή μπορεί
να ανασταλεί από τη δράση αντιοξειδωτικών παραγόντων (πχ. αμινοθειόλη ή ακετυλοκυστεϊνη
που είναι πρόδρομη ουσία της γλουταθειόνης). Η χορήγηση αυξητικών αιμοποιητικών
παραγόντων συνεισφέρει στην αντιαποπτωτική διαδικασία, χωρίς μάλιστα να έχει
καταγραφεί αύξηση της λευχαιμικής εκτροπής στους ασθενείς που έλαβαν τέτοιους
παράγοντες.[11,12]


Κλινικοεργαστηριακά
ευρήματα ΜΔΣ
Τα ΜΔΣ της παιδικής ηλικίας, λαμβάνοντας υπόψη την μερική αναθεώρηση των κριτηρίων
κατά FAB, ταξινομούνται ανάλογα με τα μορφολογικά ευρήματα στο περιφερικό αίμα
και στο μυελό των οστών σε 5 τύπους (πίνακας 5). Να σημειωθεί ότι η CMML, όπως
και η JMML, επειδή συγκεντρώνουν χαρακτηριστικά και των δύο κατηγοριών από πολλούς
ερευνητές καταγράφονται ως ανεξάρτητες οντότητες που συνιστούν ενδιάμεση ομάδα
μεταξύ των ΜΔΣ και ΜΥΣ (bridging disorders). Η κλινική εικόνα των ΜΔΣ ποικίλλει
και εξαρτάται από τον τύπο και τη βιολογική συμπεριφορά του κάθε νοσήματος.
Συνήθως προβάλλουν με συμπτώματα και ευρήματα που οφείλονται στην κυτταροπενία
μίας ή περισσοτέρων σειρών στο περιφερικό αίμα (ωχρότητα, αιμορραγική διάθεση,
συχνές λοιμώξεις μεμονωμένα ή σε συνδυασμό). Σπάνια συνυπάρχει ηπατοσπληνομεγαλία.
Ο παρακλινικός έλεγχος και, κυρίως, το επίχρισμα του περιφερικού αίματος, το
μυελόγραμμα και η οστεομυελική βιοψία, συμβάλλει τα μέγιστα στην αρχική διάγνωση
και στην περαιτέρω ταξινόμηση των ΜΔΣ.[13] Ο μυελός των οστών κατά κανόνα εμφανίζει
φυσιολογική ή αυξημένη κυτταρικότητα και ίνωση. Επιπρόσθετα, εμφανίζονται σε
μία, δύο ή και τις τρεις μυελικές σειρές μορφολογικές διαταραχές όπως: δυσερυθροποιητικές
αλλοιώσεις (υπερλόβωση και υπερκατάτμηση πυρήνων, πολλαπλοί πυρήνες, πρωτοπλασματικές
γέφυρες), μείωση κοκκίωσης των προγονικών σειρών της κοκκιώδους σειράς, παρουσία
σιδήρου γύρω από τους πυρήνες των ερυθροβλαστών (δακτυλιοειδείς σιδηροβλάστες
ως αποτέλεσμα διαταραχής των μιτοχονδρίων), άωρες μορφές και των τριών αιμοποιητικών
σειρών, παρουσία βλαστών μέχρι και 30% των εμπυρήνων κυττάρων, διαταραχή της
αρχιτεκτονικής δομής του μυελού των οστών και συνάθροιση στο κέντρο των μυελοχώρων
προμυελοκυττάρων και βλαστών γνωστών ως ALIP (Abnormal Localization of Immature
Precursors). Το ποσοστό των CD34+ κυττάρων και της ALIP συνδέεται ευθέως ανάλογα
με την πρόγνωση των ασθενών.[14]
Θεραπεία
ΜΔΣ
Όπως αναφέρθηκε προηγουμένως, ένα ποσοστό των ΜΔΣ που φτάνει περίπου το 40%
εμφανίζει δυσμενή εξέλιξη και μπορεί να καταλήξει σε οξεία λευχαιμία, ενώ μικρότερο
ποσοστό παρουσιάζει αυτόματη ύφεση (συνήθως η RA). Κακοί προγνωστικοί δείκτες
είναι η παρουσία επιθετικού τύπου ΜΔΣ (RAEB, RAEB-t), η υψηλή τιμή HbF (>10%),
η μεγάλου βαθμού θρομβοπενία (<40.000/μl), το αυξημένο ποσοστό βλαστών (20-30%),
η κυτταροπενία και των τριών σειρών, η απουσία του HLA-DR[15] και οι σύνθετες
καρυοτυπικές βλάβες.[15,16] Συνεπώς, η θεραπευτική προσέγγιση των παιδικών ΜΔΣ
εξαρτάται από τους επιμέρους προγνωστικούς δείκτες. Έτσι, είναι δυνατό σε μερικές
περιπτώσεις RA ή και RAEB να προκριθεί η απλή παρακολούθηση και οι μεταγγίσεις
αίματος, ενώ αν αποφασιστεί θεραπεία η καταλληλότερη είναι αυτή που οδηγεί στην
εκρίζωση του παθολογικού κυτταρικού κλώνου, δηλαδή η αλλογενής ΜΜΟ.[17] Επειδή,
όμως, η ΜΜΟ έχει ένα ποσοστό θνητότητας και δεν είναι πάντα δυνατή η ανεύρεση
απολύτως συμβατού δότη, κατά καιρούς έχουν χορηγηθεί εναλλακτικά σχήματα με
άλλοτε άλλα αποτελέσματα. Το ρετινοϊκό οξύ, οι ιντερφερόνες-α και -γ, η κορτιζόνη,
η αντιθυμοκυτταρική σφαιρίνη, οι αυξητικοί αιμοποιητικοί παράγοντες έχουν χρησιμοποιηθεί
αλλά τα αποτελέσματα δεν είναι πολύ ικανοποιητικά.[18,19] Η θαλιδομίδη, η κυκλοσπορίνη,
η χορήγηση χαμηλών δόσεων Ara-C εμφανίζουν ανταπόκριση σε αρκετούς ασθενείς.[20]
Νεότερα ανοσοκατασταλτικά φάρμακα, όπως το χιμαιρικό μονοκλωνικό αντίσωμα αντι-TNF
και το μονοκλωνικό αντίσωμα αντί-CD33 φαίνεται να υπόσχονται πολλά.[21]
Κλινικοεργαστηριακά
ευρήματα ΜΥΣ
Τα ΜΥΣ των παιδιών ταξινομούνται επίσης σε 5 τύπους (πίνακας 1) εκ των οποίων
η ιδιοπαθής μυελοϊνωση δεν απαντάται στην παιδική ηλικία. Στα γενικά και κοινά
χαρακτηριστικά τους περιλαμβάνονται η αύξηση, μεμονωμένα ή σε συνδυασμό, των
τριών κυτταρικών σειρών στο περιφερικό αίμα, ενώ σταθερό εύρημα σε όλους τους
τύπους αποτελεί η σπληνομεγαλία. Η κλινική εικόνα των νοσημάτων αυτών ποικίλλει
και εξαρτάται από τον τύπο και τη βιολογική συμπεριφορά του ΜΥΣ. Συνήθως προβάλλουν
με συμπτώματα και ευρήματα που οφείλονται στην υπέρμετρη ανάπτυξη μίας ή περισσοτέρων
σειρών στο περιφερικό αίμα (πχ. κακουχία, αρθραλγίες, λεμφαδενοπάθεια, δερματικά
οζίδια, σπασμοί, συχνές λοιμώξεις, μεμονωμένα ή σε συνδυασμό). Ο παρακλινικός
έλεγχος, και κυρίως το επίχρισμα του περιφερικού αίματος, το μυελόγραμμα και
η οστεομυελική βιοψία, συμβάλλει τα μέγιστα στην αρχική διάγνωση και στην περαιτέρω
ταξινόμηση των ΜΥΣ. Ο μυελός των οστών κατά κανόνα είναι υπερκυτταρικός και
συνήθως εμφανίζει υπέρμετρη ανάπτυξη μίας εκ των 3 σειρών.[22]


Μυελομονοκυτταρική
λευχαιμία νεανικού τύπου (JMML)
Η JMML προσβάλλει συνήθως αγόρια (αναλογία αγοριών προς κορίτσια 3:1) ηλικίας
0-4 χρονών και μπορεί να προβάλλει με πυρετό, επιμένουσες λοιμώξεις, αιμορραγικές
εκδηλώσεις, γενικευμένη λεμφαδενοπάθεια, χαρακτηριστικό εξάνθημα προσώπου ή
και έκζεμα. Στο περιφερικό αίμα διαπιστώνεται μετρίου βαθμού λευκοκυττάρωση,
παρουσία δυσπλαστικών κοκκιοκυττάρων, μικρός αριθμός βλαστών, αυξημένος αριθμός
μονοκυττάρων, εμπύρηνα ερυθρά αιμοσφαίρια, ήπια αναιμία και θρομβοπενία. Τα
επίπεδα της HbF είναι συνήθως υψηλά, ενίοτε ξεπερνούν και το 70%. Ο μυελός των
οστών είναι υπερκυτταρικός, εμφανίζει υπερπλασία της μυελικής σειράς με μείωση
του αριθμού των μεγακαρυοκυττάρων. Ο καρυότυπος των μυελικών κυττάρων είναι
σε ποσοστό άνω του 50% φυσιολογικός, ενώ η συχνότερη ανωμαλία που ανιχνεύεται
είναι η μονοσωμία 7. Η διάγνωση της JMML στηρίζεται σε συγκεκριμένα μείζονα
και ελάσσονα κλινικοεργαστηριακά διαγνωστικά κριτήρια (πίνακας 6). Από πειραματικά
δεδομένα προκύπτει ότι τα προγονικά κύτταρα της κοκκιώδους σειράς εμφανίζουν
αυξημένη ευαισθησία στον παράγοντα GM-CSF, χωρίς να διαπιστώνεται παθολογική
αύξηση των επιπέδων του συγκεκριμένου παράγοντα στον ορό.[23] Η συσχέτιση της
νόσου με τη νευροϊνωμάτωση τύπου Ι (NF1) έχει επιπλέον συνεισφέρει στην διερεύνηση
της παθογένειας της νόσου. Χαρακτηριστικά αναφέρεται ότι η επίπτωση της JMML
σε άτομα με NF1 είναι 350 φορές μεγαλύτερη σε σχέση με την επίπτωση σε άτομα
χωρίς NF1. Το γονίδιο NF1 έχει βρεθεί ότι κωδικοποιεί πρωτεϊνη που εμπλέκεται
στην ενδοκυττάρια μεταγωγή σήματος μέσω της οδού του ογκογονιδίου ras. Σε υγιή
άτομα το γονίδιο NF1 δρα ανασταλτικά στην οδό μεταβίβασης σήματος του ras, ενέχοντας
ιδιότητες ογκοκατασταλτικού γονιδίου.[24,25]
Ποικιλία θεραπευτικών σχημάτων έχει δοκιμαστεί στην προσπάθεια επίτευξης ύφεσης
της νόσου, όμως μόνο η ΜΜΟ έχει επιτύχει σε σχετικά ικανοποιητικό ποσοστό μακροχρόνια
ύφεση της νόσου (πενταετής επιβίωση 40%). Σε περίπτωση εκσεσημασμένης σπληνομεγαλίας
συνιστάται σπληνεκτομή πριν τη ΜΜΟ. Ευνοϊκοί προγνωστικοί δείκτες για ασθενείς
που δεν πρόκειται να υποβληθούν σε ΜΜΟ θεωρούνται η τιμή των αιμοπεταλίων >40.000/μl,,
η ηλικία κάτω των 2 ετών και τα επίπεδα της HbF <15%. Ενθαρρυντικά αποτελέσματα
έχουν διαπιστωθεί μετά χορήγηση υδροξυουρίας, ανταγωνιστών των GM-CSF αναλόγων
και νεότερων παραγώγων του ρετινοϊκού οξέος.[23]
Χρόνια
μυελογενής λευχαιμία τύπου ενηλίκου (CML)
Η CML αποτελεί το 2-5% του συνόλου της παιδικής λευχαιμίας με ετήσια επίπτωση
<1/105 περιστατικά σε άτομα μικρότερα των 20 χρόνων. Η αιτιολογία της νόσου
παραμένει άγνωστη, αν και έχει συσχετιστεί με την έκθεση σε ιονίζουσα ακτινοβολία.
Χαρακτηριστικό κυτταρογενετικό εύρημα αποτελεί ή μετάθεση t(9;22) γνωστή ως
χρωμόσωμα Ph, η οποία με τις κλασσικές κυτταρογενετικές μεθόδους ανιχνεύεται
στο 90%, ενώ με μοριακούς ανιχνευτές στο 100% των πασχόντων. Με τη μετάθεση
αυτή γίνεται σύντηξη των περιοχών bcr/abl και σχηματίζεται παθολογική πρωτεϊνη
η οποία εμφανίζει χαρακτηριστικά ενεργοποιημένης κινάσης της τυροσίνης. Η κύρια
δράση αυτής της κινάσης είναι η φωσφορυλίωση σειράς κυτταρικών πρωτεϊνών, ανάμεσα
στις οποίες συγκαταλέγονται και μόρια υπεύθυνα για τον πολλαπλασιασμό και τη
διαφοροποίηση του κυττάρου. Τελικό αποτέλεσμα όλης αυτής της διαδικασίας είναι
η μεταγωγή προς τον πυρήνα του κυττάρου συνεχούς σήματος που οδηγεί σε μη ελεγχόμενο
πολλαπλασιασμό και εκτροπή του σε κακόηθες. Η συμπτωματολογία εξαρτάται από
την παθογένεια της νόσου που οδηγεί σε υπέρμετρη αύξηση των άωρων και ώριμων
κοκκιοκυττάρων και τη συνυπάρχουσα σπληνομεγαλία. Πυρετός, ιδρώτες, κακουχία,
κοιλιακό άλγος, οστικά άλγη ή αρθραλγίες, δερματικά οζίδια λόγω εξωμυελικής
αιμοποίησης, αποτελούν τα συχνότερα εμφανιζόμενα συμπτώματα. Στα παιδιά παρουσιάζεται
συχνότερα λευκόσταση λόγω του αυξημένου αριθμού των λευκών, με αποτέλεσμα την
εκδήλωση σπασμών ή πριαπισμού. Ο αριθμός των λευκών συχνά υπερβαίνει τις 100.000/μl,
σπανιότερα ανέρχεται και άνω του 1.000.000/μl, ενώ το επίχρισμα του περιφερικού
αίμα περιλαμβάνει κύτταρα της μυελικής σειράς σε όλες τις βαθμίδες ωρίμανσης.
Στο μυελό των οστών διαπιστώνεται υπεροχή της λευκής σειράς, αύξηση των μονοπύρηνων
και παρουσία βλαστικών κυττάρων που στη χρόνια φάση της νόσου είναι <5%.
Η δραστηριότητα της αλκαλικής φωσφατάσης των λευκών είναι ελαττωμένη. Αναγκαία
και ικανή συνθήκη για τη διάγνωση της νόσου αποτελεί η ανεύρεση της μετάθεσης
t(9;22), η οποία συχνά ανιχνεύεται και σε περιόδους ύφεσης της CML.[22] Η διαφορική
διάγνωση της CML περιλαμβάνει κυρίως τη JMML, αλλά και νοσήματα όπως η ΟΜΛ και
οι διαφόρου αιτιολογίας λευχαιμοειδείς αντιδράσεις που συναντώνται επί λοιμώξεων,
συγγενών καρδιακών νοσημάτων κλπ. Στον Πίνακα 7 καταγράφονται τα χαρακτηριστικά
που διαφοροποιούν τη CML από τη JMML.
Η νόσος εμφανίζεται στη χρόνια φάση της η οποία διαρκεί περίπου 3 χρόνια. Η
επιταχυνόμενη φάση της CML χαρακτηρίζεται από ανθεκτικότητα των λευχαιμικών
κυττάρων στη θεραπεία, αύξηση του μεγέθους του σπλήνα, αναιμία και απώλεια βάρους.
Κατά κανόνα ακολουθεί η βλαστική κρίση που μπορεί να προέρχεται από κύτταρο
της μυελικής ή της λεμφικής σειράς. Τις προηγούμενες δεκαετίες η θεραπεία στη
χρόνια φάση στηριζόταν στη χορήγηση βουσουλφάνης, η οποία λόγω σημαντικών παρενεργειών
(μυελοτοξικότητα, στείρωση, δευτεροπαθείς κακοήθειες) έχει σχεδόν εγκαταλειφθεί.
Τα τελευταία χρόνια έχει δοκιμαστεί με καλύτερα αποτελέσματα η υδροξυουρία,
η ιντερφερόνη-α, η Ara-C και πρόσφατα η SΤΙ 571 (Glevec). Πάντως ως θεραπεία
εκλογής στους παιδιατρικούς ασθενείς παραμένει η ετερόλογη ΜΜΟ με πιθανότητα
πενταετούς επιβίωσης 80%.[26]
Ιδιοπαθής θρομβοκυτταραιμία
(ΕΤ)
Σπανιότερο νόσημα, καθώς μόνο 50 περιπτώσεις έχουν καταγραφεί σε παιδιά στη
διεθνή βιβλιογραφία, είναι η ΕΤ που χαρακτηρίζεται από επιμένουσα θρομβοκυττάρωση
για την οποία έχει αποκλειστεί οποιαδήποτε άλλη αιτιολογία όπως σιδηροπενία,
stress, χρόνια φλεγμονή, λοίμωξη, νόσημα του συνδετικού ιστού, νεοπλασία ή άλλη
μυελοϋπερπλαστική διαταραχή (αληθής πολυκυτταραιμία, μυελοϊνωση). Τα διαγνωστικά
κριτήρια της ΕΤ καταγράφονται στον πίνακα 8. Τα παιδιά συνήθως είναι ασυμπτωματικά
και η διάγνωση τίθεται στα πλαίσια διερεύνησης σπληνομεγαλίας που εντοπίζεται
σε τυχαία αντικειμενική εξέταση, όπως στην περίπτωση των δικών μας ασθενών,
ενώ σπανιότερα παρουσιάζουν είτε θρομβωτικά είτε αιμορραγικά επεισόδια αλλά
ποτέ και τα δύο. Συμπτώματα όπως κεφαλαλγία, παραισθησίες, αισθητικές διαταραχές
οφείλονται σε μικροαγγειακά έμφρακτα, ενώ σοβαρά θρομβωτικά επεισόδια και σπασμοί
είναι σπάνιες εκδηλώσεις στα παιδιά. Στο περιφερικό αίμα παρατηρείται επιμένουσα
θρομβοκυττάρωση που συνήθως ξεπερνά το 1.000.000/μl χωρίς διαταραχές στις υπόλοιπες
σειρές. Συχνά η μορφολογία των αιμοπεταλίων είναι διαταραγμένη (μειωμένη κοκκίωση,
δυσμορφία λοβών, μειωμένος μέσος όγκος, γιγάντιαιες μορφές) και καταγράφονται
παθολογικές τιμές στις δοκιμασίες συσσώρευσης. Ο μυελός των οστών είναι συνήθως
υπερκυτταρικός, με υπερπλασία των μεγακαρυοκυττάρων, απουσία ίνωσης και φυσιολογικές
τις υπόλοιπες σειρές. Οι χρωμοσωμικές βλάβες είναι εξαιρετικά σπάνιες, ενώ το
χρωμόσωμα Ph πάντα απουσιάζει. Η παθογένεια σχετίζεται με διαταραχή στην αλληλεπίδραση
της θρομβοποιητίνης με τον κυτταρικό της υποδοχέα (c-Mpl), που οδηγεί σε μειωμένη
κάθαρση και αυξημένη λειτουργική δραστηριότητα της θρομβοποιητίνης.[27]
Θεραπευτικά, οι αντιαιμοπεταλιακοί παράγοντες όπως η διπυριδαμόλη και η ασπιρίνη
βοηθούν στην πρόληψη θρομβώσεων, όμως η ασπιρίνη παρατείνει το χρόνο πήξης και
είναι δυνατό να προκαλέσει αιμορραγικά επεισόδια σε ασθενείς με μυελοϋπερπλαστικές
διαταραχές. Διάφοροι χημειοθεραπευτικοί παράγοντες (ραδιενεργός φώσφορος, υδροξυουρία,
αλκυλιούντες παράγοντες) έχουν χρησιμοποιηθεί με ικανοποιητική ανταπόκριση,
πλην, όμως, έχουν συσχετιστεί με σημαντικές παρενέργειες όπως μυελοτοξικότητα
και καρκινογένεση, ιδίως λευχαιμογένεση.[28] Η ιντερφερόνη-α έχει επίσης σημαντική
δράση στη μείωση του αριθμού των αιμοπεταλίων, όμως έχει και αυτή σημαντικές
παρενέργειες (λευκοπενία, συμπτώματα γρίππης, αλωπεκία, ανορεξία, μυαλγίες),
έχει αυξημένο οικονομικό κόστος και απαιτεί συχνές υποδόριες ενέσεις. Ένα νέο
φάρμακο, η αναγκρελίδη, τείνει να καθιερωθεί ως θεραπεία εκλογής στην ΕΤ στα
παιδιά. Είναι εκλεκτικός αναστολέας της cAMP φωσφοδιεστεράσης των αιμοπεταλίων,
δεν επηρεάζει τις άλλες μυελικές σειρές και έχει πολύ καλή βιοδιαθεσιμότητα.
Σε μυελογράμματα ασθενών που λαμβάνουν το φάρμακο φαίνεται ότι δεν υπάρχει μείωση
του αριθμού των μεγακαρυοκυττάρων αλλά μείωση της διαμέτρου των μεγακαρυοκυττάρων
και αναστολή της ωρίμανσής τους. Η δόση εφόδου κυμαίνεται από 1 ως 4mg ημερησίως,
ενώ η μέση δόση μετά την ύφεση είναι 2 mg.[29] Το ποσοστό ανταπόκρισης είναι
άνω του 90%, ενώ οι παρενέργειες είναι σπάνιες και συνήθως ήπιες και παροδικές
(πονοκέφαλος, υπόταση, ναυτία, αίσθημα παλμών, κοιλιακό άλγος). Δεν έχει καταγραφεί
περιστατικό λευχαιμίας σε παιδί που έχει λάβει αναγκρελίδη.[30]
Συμπεράσματα
Από τα ευρήματα της μελέτης μας φαίνεται ότι τα ΜΔΣ στα παιδιά προβάλλουν με
συμπτώματα και σημεία κυτταροπενίας στο περιφερικό αίμα, είναι πιο συχνές οι
επιθετικές τους μορφές (RAEB, RAEB-t) απ΄ότι στους ενήλικες, συνδυάζονται συχνότερα
με κυτταρογενετικές ανωμαλίες, συχνά μεταπίπτουν σε ΟΜΛ, είναι πιο συχνά στα
αγόρια, ενώ θεραπεία εκλογής είναι η αλλογενής ΜΜΟ με αποτελέσματα αρκετά ευνοϊκά
στα πρωτοπαθή. Τα ΜΥΣ της παιδικής ηλικίας έχουν κατά κανόνα σχετικά καλή πορεία,
όμως μερικές φορές μεταπίπτουν σε λευχαιμία. Σταθερό κλινικό σημείο είναι η
σπληνομεγαλία. Πλην της CML, στα ΜΥΣ εντοπίζονται λιγότερο συχνά γενετικές διαταραχές
σε σύγκριση με τα ΜΔΣ. Η αλλογενής μεταμόσχευση ΜΜΟ αποτελεί τη θεραπεία εκλογής
στη JMML και τη CML, ενώ η αναγκρελίδη καθιερώνεται ως πρώτη επιλογή στην ΕΤ
λόγω υψηλής αποτελεσματικότητας και πολύ μικρού ποσοστού παρενεργειών.
Η ευαισθητοποίηση των παιδιάτρων στην έγκαιρη διάγνωση των ΜΔΣ και των ΜΥΣ συνεπάγεται
την έγκαιρη έναρξη θεραπείας που βελτιώνει σημαντικά τα ποσοστά επιβίωσης και
προφυλάσσει από τις επιπλοκές, μεταξύ των οποίων περιλαμβάνεται η οξεία λευχαιμία.
Τα ΜΔΣ και τα ΜΥΣ της παιδικής ηλικίας έχουν αποτελέσει πεδίο αυξημένης επιστημονικής
δραστηριοποίησης τα τελευταία χρόνια. Η καταγραφή των λίγων περιστατικών που
εκδηλώνονται σε παιδιά συμβάλλει στην κατανόηση των ιδιαιτεροτήτων τους και
θα οδηγήσει σε καλύτερα θεραπευτικά αποτελέσματα.
Myelodysplastic
and myelohyperplastic syndromes of childhood
M. Moschovi, G. Trimis, V. Touliatou, Κ. Stefanaki,, V. Kitra, F. Tzortzatou-Stathopoulou
(Ann Clin Paediatr Univ Atheniensis 2004, 51(1): 39-51)
The myelodysplastic (MDS)
and myelohyperplastic (MHS) syndromes include a diverse group of clonal disorders
of progenitor hematopoietic cell that because of their rarity have not been
studied extensively in childhood. In the present study we report the special
characteristics of MDS and MHS that were diagnosed and treated in our Unit the
last five years (1998-2002) and we review the bibliographic data.
Materials of our study consisted of 8 children (6 males-2 females) aged 2-18
years old (mean age 9 years) with MDS (6 primary-2 secondary) and 5 children
(3 males-2 females) aged 1-13 years old (mean age 8 years) with MHS. Abnormal
cytogenetics was appeared in 3/8 children with MDS, while 5/8 underwent hematopoietic
stem cell transplantation (HSCT). It is remarkable that 2/6 children with idiopathic
MDS manifested acute myeloblastic leukemia (AML) in 6 and 12 months respectively.
Death occurred in 3/8 children, the other patients achieved remission for 10
months-4 years (mean time 26 months). In patients with MHS, hydroxyurea was
given to 2/5 children, while the child with JMML underwent HSCT from histocompatible
related donor twice, because meantime he manifested acute lymphoblastic leukemia
(ALL). The children with essential thrombocythemia (ET) received anagrelide.
All the patients are in remission for 2.5-4 years (mean time 38 months).
From our findings, it seems that the MDS in children are manifested more frequently
with aggressive forms, they have abnormal cytogenetics, they are often transformed
into AML and their treatment of choice is the allogeneic HSCT. The outcome of
HSCT is quite favorable in idiopathic MDS, but it is disappointing in secondary
ones. The MHS of childhood often have a good outcome and rarely are transformed
into leukemia. A standard clinical sign is splenomegaly. Except CML, MHS have
less often abnormal cytogenetics. The allogeneic HSCT is the treatment of choice
for JMML and CML, while anagrelide is established as the drug of choice in ET
without side effects.
Key Words: myelodysplastic syndromes, myelohyperplastic syndromes, childhood, leukemia.
ΒΙΒΛΙΟΓΡΑΦΙΑ
1. Emanuel PD. Myelodysplasia and myeloproliferative disorders in childhood:
an update. Br J Haematol 1999, 105:852-63.
2. Hall GW. Cytogenetic and molecular genetic aspects of childhood myeloproliferative/myelodysplastic
disorders. Acta Haematol 2002,108:171-9.
3. Mandel K, Dror Y, Poon A, Freedman MH. A practical comprehensive classification
for pediatric myelodysplastic syndromes: The CCC System. J Pediatr Hematol Oncol
2002, 24:596-605.
4. Steensma DP, Tefferi A. The myelodysplastic syndrome(s): a perspective and
review highlighting current controversies. Leuk Res 2003, 27:95-120.
5. Luna-Fineman S, Shannon KM, Atwater SK. Myelodysplastic and myeloproliferative
disorders of childhood: a study of 167 patients. Blood 1999, 93:459-66.
6. Rosenfeld C, List A. An hypothesis for the pathogenesis of myelodysplastic
syndromes: implications for new therapies. Leukemia 2000, 14:2-8.
7. Winter SS, Hanissian GA, Harville TO, Ware RE. Tumor necrosis factor-alpha
suppresses hematopoiesis in children with myelodysplasia. Med Pediatr Oncol
1997, 28:69-77.
8. Raza A, Gezer S, Mundle S, Gao XZ, Alvi S, Borok R et al. Apoptosis in bone
marrow biopsy samples involving stromal and hematopoeitic cells in 50 patients
with myelodysplastic syndromes. Blood 1995,86:268-76.
9. Mori N, Hidai H, Yokota J, Okada M, Motoji T, Oshimi K et al. Mutations of
the p53 gene in myelodysplastic syndrome and overt leukemia. Leuk Res 1995,19:869-75.
10. Bowen D. Oxidative stress and the myelodysplastic syndromes. Leuk Res 2000,24:139-40.
11. Tehranchi R, Faadel B, Forsblom AM, Christensson B, Samuelsson J, Zhivotovsky
B et al. Granulocyte-stimulating factor inhibits spontaneous cytochrome c release
and mitochondria-dependent apoptosis of myelodysplastic syndrome hematopoietic
progenitors. Blood 2003,101:1080-6.
12. Fracchiolla N, Catena F, Novembrino C, Ippolito S, Maisonneuve P, Cortelezzi
A. Possible association between reactive oxygen metabolites and karyotypic abnormalities
in myelodysplastic syndromes. Haematologica 2003,88:594-7.
13. Sasaki H, Manabe A, Kojima S, Tsuchida M, Hayashi Y, Ikuta Y et al. Myelodysplastic
syndrome in childhood: a retrospective study of 189 patients in Japan. Leukemia
2001, 15:1713-20.
14. Verburg E, Achten R, Maes B, Hagemeijer A, Boogaerts M, De Wolf-Peeters
C et al. Additional prognostic value of bone marrow histology in patients subclassified
according to the International Prognostic Scoring System for myelodysplastic
syndromes. J Clin Oncol 2003, 21:273-82.
15. Greenberg P, Cox C, Le Beau MM. International scoring system for evaluating
prognosis in myelodysplastic syndromes. Blood 1997, 78:2079-88.
16. Belli C, Acevedo S, Bengio R, Arrossagaray G, Watman N, Rossi N et al. Detection
of risk groups in myelodysplastic syndromes. A multicenter study. Haematol 2002,
87:9-16.
17. Locatelli F, Zecca M, Niemeyer C, Angelucci E, Arcese G, Bender-Gotze C
et al. Role of allogeneic bone marrow transplantation for the treatment of myelodysplastic
syndromes in childhood. Bone Marrow Transplant 1996, 18:63-68.
18. Nagatoshi Y, Okamura J, Ikuno Y, Akamatsu M, Tasaka H. Therapeutic trial
of intensified conditioning regimen with high dose cytosine arabinoside, cyclophosphamide
and either total body irradiation or busulfan followed by allogeneic bone marrow
transplantation for myelodysplastic syndrome in children. Int J Hematol 1997,
65:269-75.
19. Visani G, Tosi P, Manfroi S, Ottaviani E, Finelli C, Cenacchi A et al. All-trans
retinoic acid in the treatment of myelodysplastic syndromes. Leuk Lymphoma 1995,
19:277-80
20. Creutzig U, Bender-Gotze C, Ritter J, Zimmermann M, Stollmann-Gibbels B,
Korholz D et al. The role of intensive AML-specific therapy in treatment of
children with RAEB and RAEB-t. Leukemia 1998,1 2:652-9.
21. Biesma DH, Van-del-Tweel JK, Verdonck LF. Immunosuppressive therapy for
hypoplastic myelodysplastic syndrome. Cancer 1997, 79:1548-51.
22. Πολυχρονοπούλου-Ανδρουλακάκη Σ. Χαρακτηριστικά των μυελοϋπερπλαστικών συνδρόμων
στην παιδική ηλικία. Παιδιατρική 2002,65:222-31.12Αη1211
23. Arico M, Biondi A, Pui C. Juvenile myelomonocytic leukemia. Blood 1997,
78:1403-12.
24. Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M et al. Homozygous
inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis
type I and malignant myeloid disorders. N Engl J Med 1997, 336:1713-20.
25. Pui L, Liu J, Gish G. Bcr/Abl oncoproteins bind directly to activators of
the Ras signaling pathway. EMBO J 1994, 13:764-72.
26. Thornley I, Perentesis JP, Davies SM, Smith FO, Champagne M, Lipton JM.
Treating children with chronic myeloid leukemia in the imatinib era: a therapeutic
dilemma. Med Pediatr Oncol 2003, 41:115-7.
27. Dror Y, Blanchette VS. Essential thrombocythaemia in children. Br J Haematol
1999, 107:691-8.
28. Sterkers Y, Preudhomme C, Lai JL, Demory JL, Caulier MT, Wattel E et al.
Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia
treated with hydroxyurea: High proportion of cases with 17p deletion. Blood
1998, 91:616-22.
29. Silverstein MN, Tefferi A. Treatment of essential thrombocythemia with anagrelide.
Semin Hematol 1999, 36:23-5.
30. Lackner H, Urban C, Beham-Schmid C, Benesch M, Kerbl R, Schwinger W. Treatment
of children with anagrelide for thrombocythemia. J Pediatr Hematol Oncol 1998,
20:469-73.
