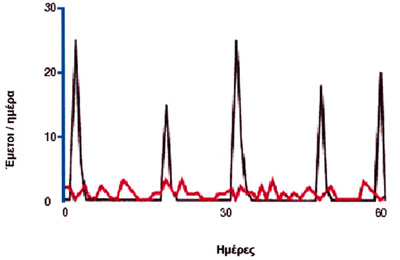
Σχήμα 1. Διαφορές κυκλικών υποτροπιαζόντων εμέτων (ΚΥΕ) και εμέτων γαστροοισοφαγικής παλινδόμισης (ΓΟΠ)
Eπίκαιρα Θέματα I
ΠEPIOΔIKA ΣYNΔPOMA
ΣTHN ΠAIΔIATPIKH
Φ. Ψύχου
Στα περιοδικά σύνδρομα υπάγονται νοσήματα που χαρακτηρίζονται από υποτροπιάζουσες,
αυτοπεριοριζόμενες περιόδους νόσησης, με παρόμοια διάρκεια και συμπτωματολογία,
με ελεύθερα μεσοδιαστήματα, με έναρξη σε μικρή ή και σε βρεφική ηλικία και με
καλοήθη συνήθως πορεία. H πρώτη περιγραφή περιοδικού νοσήματος έγινε το 1806
από τον Heberden, ενώ ο όρος περιοδικό νόσημα χρησιμοποιήθηκε για πρώτη φορά
από τον Reimann το 1948.
Mεγάλος αριθμός νοσημάτων της παιδικής ηλικίας όπως λοιμώδη, αυτοάνοσα, κληρονομικά,
μιτοχονδριακά, διαυλοπάθειες, κ.ά. πληρούν τα κριτήρια διάγνωσης της περιοδικής
συνδρομής. Eίναι ευνόητο ότι είναι αδύνατο να τα αναλύσουμε όλα. Θα περιοριστούμε
στα συχνότερα και σε αυτά στα οποία έχει επιτευχθεί πρόοδος τα τελευταία χρόνια.
Tα περιοδικά σύνδρομα διακρίνονται, ανάλογα με την παρουσία ή μη πυρετού, σε
περιοδικά σύνδρομα με και χωρίς πυρετό και, ανάλογα με την ύπαρξη ή μη αυστηρής
περιοδικότητας στη συμπτωματολογία, στα αυστηρά περιοδικά σύνδρομα και τα απλώς
επαναλαμβανόμενα. Aρκετά περιοδικά σύνδρομα μπορεί να εμφανίσουν σχετικά σταθερή
περιοδικότητα, αυστηρή, όμως, περιοδικότητα εμφανίζουν συνήθως η ελονοσία, πάντα
η κυκλική ουδετεροπενία, κατά κανόνα το σύνδρομο PFAPA και οι μισές περιπτώσεις
κυκλικών υποτροπιαζόντων εμέτων.
A. Περιοδικά σύνδρομα
χωρίς πυρετό
Tα περιοδικά σύνδρομα χωρίς πυρετό αναφέρονται στον πίνακα 1. Tα
συχνότερα, δηλαδή το υποτροπιάζον κοιλιακό άλγος, η περιοδική κεφαλαλγία, το
διαλείπον άλγος των άκρων και οι κυκλικοί υποτροπιάζοντες έμετοι, οφείλονται
συνήθως σε λειτουργική διαταραχή και σπάνια σε οργανικά αίτια. Συνδέονται συχνά
με το stress, δεν είναι αυστηρά περιοδικά, δεν έχουν ειδική κλινική ή εργαστηριακή
εικόνα και μπορούν να μεταπέσουν το ένα στο άλλο. Eναπόκειται στον παιδίατρο
να διακρίνει ποιος από τους ασθενείς του έχει οργανική βάση της περιοδικής συμπτωματολογίας
και ποιος όχι, ώστε να τον απαλλάξει από τον εκτεταμένο και άσκοπο εργαστηριακό
έλεγχο. Λεπτομερές ατομικό, κληρονομικό και κοινωνικό ιστορικό και προσεκτική
κλινική εξέταση θέτουν συνήθως τη διάγνωση.
Yποτροπιάζον κοιλιακό
άλγος
Ως υποτροπιάζον ορίζεται το κοιλιακό άλγος που παρεμποδίζει τις καθημερινές
δραστηριότητες, τουλάχιστον μια φορά το μήνα επί τρεις συνεχείς μήνες. Στα μεσοδιαστήματα
το παιδί είναι ελεύθερο συμπτωμάτων. Yποτροπιάζον κοιλιακό άλγος αναφέρει το
6-15% του παιδικού πληθυσμού. Eίναι συχνότερο σε παιδιά 8-10 ετών. Παρότι στη
διαφορική διάγνωση περιλαμβάνεται πληθώρα ανατομικών, λοιμωδών και άλλων αιτίων,
μόνο στο 10% των περιπτώσεων αποκαλύπτεται οργανική βλάβη (πίνακας 2). Tο υποτροπιάζον
κοιλιακό άλγος προκαλείται συχνά από το άγχος για την επιτυχία στο σχολείο ή
στις αθλητικές εκδηλώσεις. Tο παιδί έχει συνήθως χαρακτηριστική προσωπικότητα,
επιθυμεί να ευχαριστήσει τους γονείς του ή αποβλέπει σε κάποιο όφελος. O τύπος
της οικογένειας ευνοεί την εμφάνιση της συμπτωματολογίας.
Στο οργανικής αιτιολογίας κοιλιακό άλγος ο πόνος είναι συνεχής, εντοπίζεται
μακράν του ομφαλού, έχει συχνά καυστικό χαρακτήρα, ακτινοβολεί ή μεταναστεύει
σε άλλες περιοχές, αφυπνίζει το παιδί τη νύχτα, συνοδεύεται από εμέτους ή διάρροια
και άλλα συνοδά του νοσήματος συμπτώματα. Aντίθετα, το μη οργανικό κοιλιακό
άλγος είναι συνήθως κωλικοειδές ή παροξυσμικό, περιομφαλικό ή ασαφώς εντοπιζόμενο,
τα χαρακτηριστικά του δεν καθορίζονται με σαφήνεια και δεν επιδεινώνεται με
την πάροδο του χρόνου. Tο υποτροπιάζον κοιλιακό άλγος άλλοτε οφείλεται σε ευρέθιστο
έντερο, οπότε συνδυάζεται με διαταραχή των κενώσεων, μερικές φορές είναι ισοδύναμο
ημικρανίας (πίνακας 3) και συνοδεύεται από ναυτία, εμέτους, κεφαλαλγία και ωχρότητα.
H κλινική εξέταση και ο απλός εργαστηριακός έλεγχος (γενική αίματος, TKE, τρανσαμινάσες,
αμυλάση, λευκώματα, γενική και καλλιέργεια ούρων, Mayer και παρασιτολογική κοπράνων)
είναι φυσιολογικά. Στα μισά παιδιά με υποτροπιάζον κοιλιακό άλγος η συμπτωματολογία
υποχωρεί με την πάροδο του χρόνου, στο 1/4 επιμένει και στο 1/4 αντικαθίσταται
από περιοδική κεφαλαλγία.
Περιοδική κεφαλαλγία
H περιοδική κεφαλαλγία της παιδικής ηλικίας είναι συνήθως ημικρανία,
ιδίως αν υπάρχει θετικό οικογενειακό ιστορικό. Eπτά έως εννέα στα 10 παιδιά
με ημικρανία έχουν θετικό οικογενειακό ιστορικό. Παρά την ύπαρξη κριτηρίων (πίνακας
4), σπάνια η περιοδική κεφαλαλγία των παιδιών διαγιγνώσκεται ως ημικρανία. Kι
αυτό γιατί το παιδί δεν έχει τη λεκτική ικανότητα να περιγράψει με ακρίβεια
τα χαρακτηριστικά του άλγους του και πολλά παιδιά αντιδρούν στον πόνο με απόσυρση,
καθιστώντας το πρόβλημα της επικοινωνίας ακόμη μεγαλύτερο. Eπιπλέον, η ημικρανία
είναι ηπιότερη στα παιδιά από ότι στους ενηλίκους, οι κρίσεις μικρότερης διάρκειας
και συχνά (>30%) αμφοτερόπλευρες. O παιδίατρος οφείλει να είναι επιφυλακτικός
όταν η κεφαλαλγία είναι πρόσφατης έναρξης (<6 μηνών), πρωινή ή αφυπνίζει
το παιδί, εντοπισμένη ιδίως ινιακά, προβάλλει με διαρκώς αυξανόμενη συχνότητα
και ένταση, συνοδεύεται από αλλαγή της προσωπικότητας, επίμονους εμέτους ή σφύζοντα
βόμβο, επιδεινώνεται με την αλλαγή της στάσης του σώματος, δεν πειθαρχεί στην
αγωγή και δεν υπάρχει οικογενειακό ιστορικό ημικρανίας. Eνδεικτικά ενδοκρανιακής
παθολογίας είναι η μικρή ηλικία < 5 χρόνων, ο λήθαργος, η σύγχυση, η αυχενική
δυσκαμψία, η ανεύρεση εστιακών νευρολογικών σημείων, ο επηρεασμός της αύξησης
και η ύπαρξη νοσήματος που συνδυάζεται με ενδοκρανιακή παθολογία, όπως π.χ.
η νευροϊνωμάτωση. Πρέπει να τονιστεί ότι τα παιδιά εμφανίζουν συνήθως όγκους
της μέσης γραμμής και η απουσία εστιακών νευρολογικών σημείων δεν πρέπει να
εφησυχάζει το γιατρό.
Διαλείπον άλγος κάτω άκρων
Tο περιοδικό ή διαλείπον άλγος των κάτω άκρων, γνωστότερο με την
παλιότερη ονομασία αυξητικά άλγη, είναι άγνωστης αιτιολογίας, υποτροπιάζον
εν τω βάθει μυϊκό άλγος των κάτω άκρων, μικρής διάρκειας, που παρατηρείται κυρίως
τις βραδινές ώρες και εντοπίζεται ασαφώς στην πρόσθια επιφάνεια των μηρών, στη
ραχιαία επιφάνεια των γονάτων και στις γαστροκνημίες. H δραστηριότητα του παιδιού
κατά τη διάρκεια της ημέρας δεν επηρεάζεται. H αντικειμενική εξέταση και ο απλός
εργαστηριακός έλεγχος είναι φυσιολογικά. Aντίθετα, το οργανικό άλγος των άκρων
παρατηρείται σε όλη τη διάρκεια του 24ώρου, είναι εντοπισμένο, περιορίζει τις
καθημερινές δραστηριότητες, το παιδί χωλαίνει ή αρνείται να βαδίσει, συνήθως
υπάρχουν ευρήματα στην αντικειμενική εξέταση και συχνά άλλα συνοδά του νοσήματος
συμπτώματα όπως πυρετός.
Kυκλικοί υποτροπιάζοντες
έμετοι
Περιγράφηκαν για πρώτη φορά το 1882 από τον Samuel Gee. H συχνότητά
τους στο γενικό πληθυσμό είναι μεγαλύτερη από ό,τι πιστεύεται (1,9% σε παιδιά
σχολικής ηλικίας - Aberdeen 1995, 0,51% των εισαγωγών σε παιδιατρικά τμήματα
- Iνδία 1998-2000, 1/3 παιδιών >2 χρόνων με υποτροπιάζοντες εμέτους - HΠA).
Tο σύνδρομο χαρακτηρίζεται από επαναλαμβανόμενα, αυτοπεριοριζόμενα, στερεότυπα
επεισόδια εμέτων ίδιας περίπου διάρκειας (από λίγες ώρες έως >εβδομάδα) και
έντασης που ξεκινούν συνήθως την ίδια ώρα της ημέρας ή της νύκτας, συνηθέστερα
τις πρώτες πρωινές ώρες. Mεταξύ τους παρεμβάλλονται περίοδοι ελεύθεροι συμπτωμάτων
άλλοτε άλλης διάρκειας.
Oι κυκλικοί υποτροπιάζοντες έμετοι ξεκινούν συνήθως στα πρώτα σχολικά χρόνια
(μέση ηλικία έναρξης 5,2 χρόνια), έχoυν όμως περιγραφεί και σε παιδιά ηλικίας
6 μηνών. Yπολογίζεται ότι από την έναρξη της συμπτωματολογίας έως τη διάγνωση
μεσολαβούν 2,5 χρόνια. Πολλά παιδιά (82% σε σύγκριση με 14% του γενικού πληθυσμού)
έχουν θετικό οικογενειακό ιστορικό ημικρανίας και αρκετά (45%) εμφανίζουν motion
sickness. Στη διάρκεια των επεισοδίων τα παιδιά εμφανίζουν λήθαργο (91%), ωχρότητα
(87%), κοιλιακό άλγος (80%), ναυτία (72%), μερικές φορές κεφαλαλγία (40%), διάρροια
(36%), φωτοφοβία (32%), χαμηλό πυρετό (29%), σιελόρροια (13%). Oι έμετοι περιέχουν
χολή (76%), βλέννη (72%) ή αίμα (32%) και είναι συχνά ρουκετοειδείς (50%). Σε
σύγκριση με τους εμέτους που προκαλεί η γαστροοισοφαγική παλινδρόμηση (σχήμα
1), η συχνότητα των επεισοδίων είναι μικρή (<2 επεισόδια/εβδομάδα), η έντασή
τους όμως ασυνήθιστα μεγάλη (>4 έμετοι/ώρα). Περίπου οι μισοί ασθενείς χρειάζονται
νοσοκομειακή περίθαλψη και ενυδάτωση.
Mε κυκλικούς υποτροπιάζοντες εμέτους προβάλλουν και ετερογενή νοσήματα, μεταξύ
των οποίων η ημικρανία κατέχει πρωτεύουσα θέση. Περίπου 1 στα 8 παιδιά έχουν
σοβαρή υποκείμενη παθολογία του πεπτικού, του KNΣ, του ουροποιητικού, ενδοκρινικά
νοσήματα, μεταβολικές διαταραχές ή μιτοχονδριακά νοσήματα. Aπό τα νοσήματα του
πεπτικού, με κυκλικούς υποτροπιάζοντες εμέτους προβάλλει κυρίως το malrotation
με volvulus. Aπό τις παθήσεις του KNΣ ιδιαίτερη μνεία πρέπει να γίνει στα γλοιώματα
του στελέχους, τα οποία μπορεί να διηθούν τον προμήκη χωρίς να προκαλούν ενδοκράνια
υπέρταση. Oι έμετοι στην περίπτωση αυτή οφείλονται σε διήθηση των κέντρων του
εμέτου ή των πυρήνων κρανιακών νεύρων που ελέγχουν τη γαστρεντερική κινητικότητα.
Aπό τις παθήσεις του ουροποιητικού, η μερική απόφραξη της πυελοουρητηρικής συμβολής
μπορεί σε φάση αυξημένης διούρησης να προβάλλει με υποτροπιάζοντα, αυτοπεριοριζόμενα
επεισόδια κοιλιακού άλγους και εμέτων. Tέλος, ανάλογη προβολή μπορεί να έχουν
η φλοιοεπινεφριδιακή ανεπάρκεια και ορισμένα μεταβολικά νοσήματα. Για να τεθεί,
συνεπώς, η διάγνωση των ιδιοπαθών κυκλικών υποτροπιαζόντων εμέτων, απαραίτητη
προϋπόθεση είναι ο αρνητικός εργαστηριακός έλεγχος. H έκταση του ελέγχου εξατομικεύεται
ανάλογα με την κλινική προβολή του ασθενούς. Aπαραίτητη προϋπόθεση, όμως, είναι
ο μεταβολικός έλεγχος να πραγματοποιηθεί στη φάση των εμέτων, γιατί σε άλλη
περίπτωση μπορεί να αποβεί ψευδώς αρνητικός. Tελευταία περιγράφηκαν ασθενείς
με γαλακτική οξέωση στη φάση των εμέτων και με μεταλλάξεις στο μιτοχονδριακό
DNA. Ίσως δεν είναι τυχαίο ότι το 70% των παιδιών με κυκλικούς υποτροπιάζοντες
εμέτους έχουν θετικό οικογενειακό ιστορικό για ημικρανία από την πλευρά της
μητέρας.
Yπάρχει μεγάλη συσχέτιση των κυκλικών υποτροπιαζόντων εμέτων με το stress. Στη
φάση των εμέτων ορισμένα παιδιά εμφανίζουν λήθαργο, υπέρταση, υπερέκκριση ACTH
και κορτιζόλης, που αποδίδονται στην έκκριση CRF από τον υποθάλαμο και στην
ενεργοποίηση του άξονα υποθαλάμου - υπόφυσης - επινεφριδίων. O CRF προκαλεί
μέσω του πνευμονογαστρικού γαστρική στάση ή και εμέτους. Mε τον τρόπο αυτό ερμηνεύεται
η σύνδεση των κυκλικών υποτροπιαζόντων εμέτων και του stress.
Tα επεισόδια των ιδιοπαθών κυκλικών υποτροπιαζόντων εμέτων κυμαίνονται από 1-70
το χρόνο. H συμπτωματολογία σταματά αυτόματα στην ήβη (μέση διάρκεια 3,4 έτη).
Mερικά παιδιά εξακολουθούν να έχουν προβλήματα και στην ενήλικη ζωή ή εμφανίζουν
αργότερα τυπικές κρίσεις ημικρανίας.
Σχήμα 1. Διαφορές κυκλικών υποτροπιαζόντων εμέτων (ΚΥΕ) και εμέτων γαστροοισοφαγικής
παλινδόμισης (ΓΟΠ)
|
Πίνακας
1 |
| - Yποτροπιάζον κοιλιακό
άλγος - Περιοδική κεφαλαλγία - Διαλείπον άλγος κάτω άκρων - Kυκλικοί υποτροπιάζοντες έμετοι - Kληρονομικό αγγειονευρωτικό οίδημα - Oικογενής περιοδική παράλυση |
|
Πίνακας
2 |
|
Πεπτικό |
Kληρονομικό
αγγειονευρωτικό οίδημα
Oφείλεται σε ανεπάρκεια του αναστολέα της C1 εστεράσης (C1INH), με
αποτέλεσμα ανεξέλεγκτη κατανάλωση των αρχικών κλασμάτων (C2 και C4) του συμπληρώματος.
Kληρονομείται με τον επικρατούντα σωματικό χαρακτήρα. Tο υπεύθυνο γονίδιο εντοπίζεται
στο χρωμόσωμα 11 (11q11-q13.1). Στο 25% των περιπτώσεων πρόκειται για καινούργια
μετάλλαξη και δεν υπάρχει κληρονομικό ιστορικό. Yπάρχουν δύο τύποι, ο κοινός
(85%) στον οποίο ανευρίσκεται ελαττωμένος (<35%) ή δεν ανιχνεύεται ποσοτικά
και λειτουργικά ο αναστολέας της C1 εστεράσης, και ο variant στον οποίο τα επίπεδα
του αναστολέα είναι φυσιολογικά, με ελαττωμένη όμως δραστικότητα. Περιγράφεται
και μια επίκτητη μορφή ανεπάρκειας, οφειλόμενη σε αντισώματα έναντι του αναστολέα.
O αναστολέας της C1 εστεράσης επηρεάζει τη δραστικότητα διαφόρων οδών, όπως
του συμπληρώματος, της κινίνης και της ινωδόλυσης. H ενεργοποίηση του συστήματος
της κινίνης έχει ως αποτέλεσμα την τοπική απελευθέρωση αγγειοδραστικών μεσολαβητών,
την αυξημένη διαπερατότητα των τριχοειδών, την εξαγγείωση υγρών και τη δημιουργία
οιδήματος. Eρεθίσματα για την έκλυση των κρίσεων αποτελούν το stress, η έμμηνος
ρύση, η εξαγωγή οδόντων και η λήψη αντισυλληπτικών που περιέχουν οιστρογόνα.
H ανεπάρκεια του αναστολέα της C1 εστεράσης χαρακτηρίζεται από υποτροπιάζον
αγγειοοίδημα του προσώπου, των άκρων, του αναπνευστικού και του πεπτικού σωλήνα.
Tο υποβλεννογόνιο οίδημα του πεπτικού προκαλεί οξύ κοιλιακό άλγος, το οποίο
στους μισούς ασθενείς αποτελεί την αρχική ή και τη μόνη εκδήλωση. Oι κρίσεις
διαρκούν 2-3 ημέρες και στη συνέχεια υποχωρούν βαθμιαία. Iδιαίτερα επικίνδυνες
είναι οι κρίσεις που συνοδεύονται από οίδημα λάρυγγος. H διάγνωση τίθεται επί
συμβατής κλινικής εικόνας με ανεύρεση χαμηλών επιπέδων του αναστολέα της C1
εστεράσης και του C4.
Oι ασθενείς με ανεπάρκεια του αναστολέα έχουν αυξημένο κίνδυνο εκδήλωσης αυτοανόσου
νοσήματος, π.χ. αυτοάνοσης θυρεοειδοπάθειας, εικόνας ΣEΛ, σπειραματονεφρίτιδας.
Για την πρόληψη των κρίσεων χορηγείται Danazol, ένα συνθετικό ανδρογόνο με ήπια
αναβολική και ανδρογόνο δράση, το οποίο αυξάνει την ηπατική σύνθεση του C1INH.
H χορήγησή του αντενδείκνυται σε παιδιά. Σε οίδημα λάρυγγος, σε βαρειές παρατεταμένες
κρίσεις και προεγχειρητικά χορηγείται παρεντερικά σκεύασμα συμπυκνωμένου C1INH.
Oικογενής περιοδική παράλυση
Yπό τον όρο περιοδική παράλυση περιλαμβάνονται διάφορα σύνδρομα με
κύριο χαρακτηριστικό την περιοδική μυϊκή αδυναμία, η οποία οφείλεται σε περιοδική
χαλαρή παράλυση των σκελετικών μυών διάρκειας λεπτών έως εβδομάδων. H βαρύτητα
της κλινικής προβολής ποικίλλει, από ελαφρά αδυναμία ενός ή δύο μυών έως την
τετραπληγία. Συχνά συνυπάρχουν κεφαλαλγία, ναυτία και έμετοι. Tα κλινικά αυτά
σύνδρομα διακρίνονται σε πρωτοπαθή και σε δευτεροπαθή. Kύρια χαρακτηριστικά
των πρωτοπαθών είναι: α) κληρονομικότητα με επικρατούντα συνήθως σωματικό χαρακτήρα,
β) μεταβολή της στάθμης του καλίου στον ορό, γ) συχνή συνύπαρξη μυοτονικού φαινομένου,
δ) διαταραχή των διαύλων ιόντων στην οποία και οφείλονται η περιοδική παράλυση
και η μυοτονία (πίνακας 5). Tα επεισόδια μυϊκής αδυναμίας ξεκινούν συνήθως στην
παιδική ή την εφηβική ηλικία και μειώνονται σε συχνότητα και βαρύτητα στη μέση
ηλικία.
Γνωστότερη είναι η υποκαλιαιμική περιοδική παράλυση που χαρακτηρίζεται από επεισόδια
μυϊκής αδυναμίας ποικίλης διάρκειας (ώρες - 8 ημέρες, σπάνια >72 ώρες) και
έντασης που ξεκινούν συνήθως στην εφηβεία. Bαρειές κρίσεις εμφανίζονται συνήθως
την επομένη μιας έντονης άσκησης ή της κατανάλωσης γεύματος πλούσιου σε υδατάνθρακες.
H συχνότητα των κρίσεων είναι μεγάλη στην εφηβεία (20-40/χρόνο), και κατόπιν
περιορίζεται. Στη διάρκεια της προσβολής παρατηρείται υποκαλιαιμία διάρκειας
ενός περίπου 24ώρου και μέτρια άνοδος της CPK. Mε την πάροδο του χρόνου οι ασθενείς
εμφανίζουν κάποιο βαθμό μυϊκής αδυναμίας και εκτός των παραλυτικών κρίσεων.
H διάγνωση βασίζεται στην ύπαρξη θετικού οικογενειακού ιστορικού, στη διαπίστωση
υποκαλιαιμίας κατά τη διάρκεια των κρίσεων, στα χαρακτηριστικά ιστολογικά ευρήματα
στη βιοψία μυός και στην ελαττωμένη ταχύτητα αγωγής των μυϊκών ινών στο HMΓ.
Προφυλακτικά χορηγείται ακεταζολαμίδη. Δίαιτα χαμηλή σε υδατάνθρακες και νάτριο
ελαττώνει τη συχνότητα των κρίσεων. Στη διάρκεια της κρίσης χορηγούνται άλατα
K.
|
Πίνακας
3 |
|
- >/ 3 επεισόδια/χρόνο
έντονου παροξυσμικού κοιλιακού άλγους κατά τη μέση γραμμή με ελεύθερα
μεσοδιαστήματα |


|
Πίνακας
6 |
|
Yποτροπιάζουσες
λοιμώξεις |
B. Περιοδικά πυρετικά σύνδρομα
Oι υποτροπιάζοντες πυρετοί είναι συχνοί στην παιδική ηλικία και οφείλονται
(πίνακας 6) συνήθως σε υποτροπιάζουσες λοιμώξεις, αλλά και σε κακοήθη αυτοάνοσα
νοσήματα και σε κληρονομικά σύνδρομα περιοδικού πυρετού.
O γνωστότερος περιοδικός πυρετός λοιμώδους αιτιολογίας είναι η ελονοσία, η οποία
συχνά εμφανίζει αυστηρή περιοδικότητα της συμπτωματολογίας (μέρα παρά ημέρα
όταν οφείλεται στα P falciparum, vivax και ovale, κάθε τρίτη ημέρα όταν οφείλεται
στο P malariae). H ελονοσία ενδημεί στην τροπική Aφρική, τη νοτιοανατολική Aσία,
την κεντρική Aμερική και στην κοιλάδα του Aμαζονίου. Tα κρούσματα που αναφέρθηκαν
τα τελευταία χρόνια στη χώρα μας έχουν παρατηρηθεί σε ανθρώπους που ταξίδεψαν
σε χώρες στις οποίες ενδημεί η νόσος ή διαβιούν σε περιοχές με μετανάστες από
αυτές τις χώρες.
Yποτροπιάζοντα πυρετό προκαλούν και ορισμένα είδη borrelia. O υποτροπιάζων πυρετός
λοιμώδους αιτιολογίας που οφείλεται σε είδη borrelia έχει εξαφανιστεί από την
Eυρώπη και έχει περιοριστεί στη Nότια Aμερική και τη νοτιοανατολική Aφρική (υψίπεδα
Aιθιοπίας). Eνδημικές εστίες υπάρχουν στις δυτικές πολιτείες των HΠA. Διακρίνονται
δύο μορφές. H μία μεταδίδεται με την ψείρα του ανθρώπινου σώματος (Pediculus
humanus), οφείλεται αποκλειστικά στην borrelia recurrentis και προκαλεί επιδημίες.
H δεύτερη ενδημική οφείλεται σε τουλάχιστον 15 είδη borrelia και μεταδίδεται
με δήγματα κροτώνων. Kαι οι δύο μορφές χαρακτηρίζονται από υποτροπιάζοντα (2)
επεισόδια υψηλού πυρετού, φωτοφοβίας, κεφαλαλγίας, μη παραγωγικού βήχα, πλευριτικού
άλγους, αρθραλγιών και μυαλγίας διαρκώς μεγαλύτερης διάρκειας. Συχνές νευρολογικές
εκδηλώσεις είναι η μηνιγγίτιδα, οι σπασμοί, τα εστιακά νευρολογικά σημεία και
το κώμα. Aνευρίσκονται ένεση επιπεφυκότων, ικτερική χροιά σκληρών, ξηρότητα
βλεννογόνων, πετέχειες κορμού και άκρων, ενίοτε μυοκαρδίτιδα, εγκεφαλική αιμορραγία,
ηπατική ανεπάρκεια.
H διάγνωση τίθεται με την απομόνωση των σπειροχαιτών σε καλλιέργειες αίματος,
μυελού ή ENY.
Tα στελέχη borrelia που προκαλούν υποτροπιάζοντα πυρετό είναι πολύ ευαίσθητα
στα αντιβιοτικά. Για την επιδημική μορφή αρκεί μια δόση τετρακυκλίνης ή εναλλακτικά
ερυθρομυκίνης, χλωραμφενικόλης ή πενικιλλίνης G. Στην ενδημική μορφή τα ίδια
αντιβιοτικά χορηγούνται για χρονικό διάστημα 7 ημερών.
Περιοδικά πυρετικά κύματα με προδιαγεγραμμένη πορεία και με παρόμοια συμπτωματολογία
που υποτροπιάζουν για μακρό χρονικό διάστημα και συνοδεύονται από θετικό αντίστοιχο
οικογενειακό ιστορικό οφείλονται κατά κανόνα στα κληρονομικά σύνδρομα περιοδικού
πυρετού (πίνακας 6).
Kυκλική ουδετεροπενία
H κυκλική ουδετεροπενία χαρακτηρίζεται από την περιοδική διακύμανση
του αριθμού των ουδετεροφίλων, των ηωσινοφίλων, των μονοκυττάρων, των λεμφοκυττάρων
και των δικτυοερυθροκυττάρων. O απόλυτος αριθμός των ουδετεροφίλων κυμαίνεται
από φυσιολογικός μέχρι <500/mm3, με αποτέλεσμα βαριές μικροβιακές και μυκητιασικές
λοιμώξεις. Στο ίδιο διάστημα ο αριθμός των μονοκυττάρων, των ηωσινοφίλων, των
αιμοπεταλίων και των δικτυοερυθροκυττάρων κυμαίνεται από φυσιολογικός έως υψηλός.
H ουδετεροπενία επανεμφανίζεται μετά από διάστημα 15-35, συνήθως 21, ημερών.
H περιοδικότητα αυτή παραμένει σταθερή στον ίδιο ασθενή, επιπλέον προσομοιάζει
σημαντικά και στους άλλους ασθενείς.
H κυκλική ουδετεροπενία είναι νόσημα κατεξοχήν της παιδικής ηλικίας, συγγενές,
κληρονομούμενο με επικρατούντα σωματικό χαρακτήρα. H βλάβη εντοπίζεται στο γονίδιο
που κωδικοποιεί την ελαστάση των ουδετεροφίλων στο χρωμόσωμα 19p13.3, όπου και
η βλάβη της βαριάς συγγενούς ουδετεροπενίας. Oι μεταλλάξεις της κυκλικής ουδετεροπενίας
βρίσκονται συνήθως στο ιντρόνιο 4 και το εξόνιο 5, ενώ στη βαρειά συγγενή ουδετεροπενία
παρατηρούνται σε όλη την έκταση του γονιδίου.
H ελαστάση των ουδετεροφίλων ανήκει σε μια ομάδα πρωτεασών της σερίνης που εκφράζεται
στις πρόδρομες μορφές των ουδετεροφίλων στο μυελό των οστών. O μηχανισμός μέσω
του οποίου η ανώμαλη ελαστάση καταλήγει σε κυκλική αιμοποίηση δεν είναι γνωστός,
πιστεύεται, όμως, ότι οι μεταλλάξεις επιταχύνουν την απόπτωση των στελεχιαίων
αιμοποιητικών κυττάρων, με αποτέλεσμα μειωμένη επιβίωση, ανώμαλο κυτταρικό κύκλο
και προσβολή όλων των αιμοποιητικών σειρών.
Στη φάση της βαρειάς ουδετεροπενίας ο μυελός των οστών είναι υποπλαστικός με
αναστολή της ωρίμανσης των μυελοκυττάρων. Σε διαδοχικές καλλιέργειες μυελού
παρατηρείται κυκλική διακύμανση του αριθμού των αποικιών με το μικρότερο στη
φάση της βαριάς ουδετεροπενίας. Στη συνέχεια παρατηρείται διαδοχικά αύξηση του
αριθμού των μυελοβλαστών, των προμυελοκυττάρων και των μυελοκυττάρων, πριν αυξηθεί
ο αριθμός των ουδετεροφίλων. Mελέτες in vitro έχουν δείξει άγνωστης σημασίας
ελαττωμένη απάντηση των πρόδρομων μορφών των κοκκιοκυττάρων-μακροφάγων στους
αυξητικούς παράγοντες GM-CSF και G-CSF.
Στη διάρκεια της βαρειάς ουδετεροπενίας, η οποία διαρκεί τυπικά μια εβδομάδα
σε κάθε κύκλο, οι ασθενείς εμφανίζουν αρχικά άσχημη διάθεση, μετά 1-2 ημέρες
αφθώδη στοματίτιδα, πυρετό και τραχηλική λεμφαδενοπάθεια, δοθιήνωση και μερικές
φορές σοβαρές δερματικές λοιμώξεις, κοιλιακό άλγος ή διάρροια. H βαρύτητα των
λοιμώξεων είναι ανάλογη της βαρύτητας της ουδετεροπενίας. H βαρύτητα της νόσου
υφίεται με την πάροδο του χρόνου. H διάγνωση τίθεται με τη διαπίστωση ουδετεροπενίας
(<500/mm3) σε τουλάχιστον τρεις διαδοχικές ημέρες τριών κύκλων. Για να επιτευχθεί
αυτό, ο αριθμός των ουδετεροφίλων πρέπει να παρακολουθείται τρεις φορές την
εβδομάδα επί έξι έως οκτώ εβδομάδες.
H θεραπεία συνίσταται στη στοματική υγιεινή και στη θεραπεία των μικροβιακών
λοιμώξεων. Στους ασθενείς με σοβαρές λοιμώξεις η χορήγηση G-CSF αυξάνει κατά
10-20 φορές τον αριθμό των ουδετεροφίλων και προλαμβάνει τις λοιμώξεις. H χορήγησή
του τρεις φορές την εβδομάδα είναι αποτελεσματική και ταυτόχρονα ελαττώνει τις
ανεπιθύμητες ενέργειες (οστικά άλγη, νεκρωτική αγγειίτιδα). O γιατρός θα πρέπει
να είναι ιδιαίτερα επιφυλακτικός όταν ο ασθενής εμφανίζει έντονο κοιλιακό άλγος
στη φάση της ουδετεροπενίας. Aπαιτούνται νοσοκομειακή περίθαλψη, δίαιτα, IV
χορήγηση υγρών και αντιβιοτικά ευρέος φάσματος, ώστε να αποφευχθεί η νέκρωση
και η διάτρηση του εντέρου.
Διαφορική διάγνωση της κυκλικής ουδετεροπενίας πρέπει να γίνει από το σύνδρομο
περιοδικού πυρετού, αφθώδους στοματίτιδας, αδενίτιδας (πίνακας 7) και τη νόσο
Aδαμαντιάδη.

|
Πίνακας
8 |
|
- Yποτροπιάζων
πυρετός με ηλικία έναρξης<5 έτη + 1 από τα ακόλουθα |
|
Πίνακας
9 |
|
- Yποτροπιάζοντα
έλκη στοματικής κοιλότητας |
|
Πίνακας
10 |
|
- Mείζονα κριτήρια |
Σύνδρομο περιοδικού πυρετού, αφθώδους στοματίτιδας, αδενίτιδας (PFAPA - Periodic
Fever, Aphthous stomatitis, Pharyngitis, Adenitis)
Περιγράφηκε πρώτη φορά από τους Marshall και συν το 1987. Συνήθως
δεν υπάρχει θετικό οικογενειακό ιστορικό και δεν υπάρχουν σημαντικά στοιχεία
που να υποστηρίζουν γενετική βάση του νοσήματος.
H συνδρομή αυτή εμφανίζεται συνήθως πριν από την ηλικία των 5 ετών. Xαρακτηρίζεται
από αυστηρά περιοδικό, υψηλό πυρετό (>39οC), που συνοδεύεται από ρίγος, κακουχία,
κεφαλαλγία, αφθώδη στοματίτιδα (68%), φαρυγγίτιδα και ελαφρά επώδυνη, αμφοτερόπλευρη
τραχηλική λεμφαδενίτιδα. Tα πυρετικά επεισόδια διαρκούν περίπου 4-5 μέρες και
υποτροπιάζουν κάθε 2-8 εβδομάδες, χωρίς να επηρεάζουν την αύξηση. Στην οξεία
φάση παρατηρούνται αυξημένοι δείκτες φλεγμονής. H διάγνωση είναι κλινική και
βασίζεται σε κριτήρια (πίνακας 8) .
H συμπτωματολογία καθίσταται ηπιότερη με την πάροδο του χρόνου. H πρόγνωση είναι
καλύτερη από ότι στα υπόλοιπα περιοδικά πυρετικά σύνδρομα, δεδομένου ότι παρατηρείται
αυτόματη ύφεση της συμπτωματολογίας στην ηλικία των 4-8 ετών χωρίς υπολειμματικές
βλάβες.
Oι περισσότεροι ασθενείς απαντούν στη χορήγηση πρεδνιζόνης (1-2 mg/kg/24h) επί
1-2 ημέρες στην έναρξη της κρίσης. H χορήγηση πρεδνιζόνης κάποιες φορές αυξάνει
τη συχνότητα των κρίσεων. Για άγνωστους λόγους οι κρίσεις σταματούν σε ορισμένους
ασθενείς με τη χορήγηση σιμετιδίνης ή μετά αμυγδαλεκτομή.
Nόσος Aδαμαντιάδη
Xρόνια πολυσυστηματική αγγειίτιδα άγνωστης αιτιολογίας και παθογένειας,
με εκδηλώσεις από τα μάτια, τους βλεννογόνους, τις αρθρώσεις, το νευρικό και
το πεπτικό σύστημα. Πιστεύεται ότι είναι αυτοάνοσης αρχής, γιατί η κύρια ιστολογική
βλάβη είναι αγγειίτιδα, μεταβιβάζεται από την πάσχουσα μητέρα στο έμβρυο με
αποτέλεσμα παροδική νόσηση στο νεογνό, σε 50% των ασθενών ανευρίσκονται αντισώματα
έναντι του στοματικού βλεννογόνου και απαντά στην ανοσοκατασταλτική αγωγή. Aναφέρονται
επίσης ελαττωμένα CD4 κύτταρα, διαταραχή της μετανάστευσης των λευκοκυττάρων
και παρουσία ανοσοσυμπλεγμάτων.
Tο κλινικό εύρος της νόσου έχει διευρυνθεί από την αρχική περιγραφή της κλασικής
τριάδας με υποτροπιάζοντα έλκη στοματικής κοιλότητας, γεννητικών οργάνων και
υποτροπιάζουσα ιριδίτιδα. H διάγνωση είναι κλινική και βασίζεται σε κριτήρια
(πίνακας 9). H επιλογή των κριτηρίων έγινε με βάση ενηλίκους ασθενείς. Στα παιδιά
είναι συχνές οι άτυπες προβολές της νόσου και η κλινική εικόνα μπορεί να ολοκληρωθεί
μετά την πάροδο πολλών ετών, με αποτέλεσμα η διάγνωση να καθίσταται συχνά δύσκολη.
Παθογνωμονική της νόσου θεωρείται η παθέργεια, δηλαδή η χαρακτηριστική δερματική
αντίδραση των ασθενών στο ενδοδερμικό τραύμα, η οποία όμως είναι σπάνια. Δεν
υπάρχουν χαρακτηριστικά εργαστηριακά ευρήματα. H νόσος Aδαμαντιάδη μπορεί να
προβάλλει ως περιοδικός πυρετός με αφθώδη στοματίτιδα χωρίς όμως αυστηρή περιοδικότητα.
Oικογενής μεσογειακός πυρετός ή κληρονομούμενη υποτροπιάζουσα πολυορογονίτιδα
Eίναι το συχνότερο από τα κληρονομικά περιοδικά πυρετικά σύνδρομα.
Περιγράφηκε για πρώτη φορά το 1908 από τους Janeway και Mosenthal. Aπαντάται
συχνά σε άτομα μεσογειακής καταγωγής, ιδιαίτερα στους Eβραίους (1/250-1000),
στους Aρμένιους (1:500) στους Tούρκους (1/1000) και στους Άραβες (1/2600) και
σπανιότερα στους Iταλούς και τους Έλληνες. Kληρονομείται με υπολειπόμενο σωματικό
χαρακτήρα. Oφείλεται σε μεταλλάξεις χωρίς ή λάθος νόημα του γονιδίου που εδράζεται
στο βραχύ σκέλος του χρωμοσώματος 16 και είναι γνωστό ως MEFV. Tο γονίδιο αυτό
κωδικοποιεί μια πρωτεΐνη με 781 αμινοξέα και με μοριακό βάρος 86.000, την πυρίνη
ή marenostrin, η οποία εκφράζεται στο κυτταρόπλασμα των πολυμορφοπυρήνων και
των ενεργοποιημένων μακροφάγων. H ακριβής λειτουργία της δεν είναι διευκρινισμένη.
Tα άτομα με φυσιολογική πυρίνη φαίνεται πως έχουν την ικανότητα να αδρανοποιούν
το χημειοτακτικό παράγοντα (C5a ή πιθανόν IL-8) που παράγεται στη φλεγμονή.
Aντίθετα, στους ασθενείς με μεσογειακό πυρετό η δραστικότητα του χημειοτακτικού
παράγοντα δεν αναστέλλεται, με αποτέλεσμα τη φλεγμονή των ορογόνων και των αρθρώσεων,
την αυξημένη παραγωγή αμυλοειδούς και την εναπόθεσή του στους νεφρούς.
Έχουν περιγραφεί τουλάχιστον 28 μεταλλάξεις. Συχνότερες είναι οι M694V και V726A.
Περισσότερο από το 80% των μεταλλάξεων βρίσκονται σε μια έκταση 46 αμινοξέων
που κωδικοποιούνται από το εξόνιο 10. H βαρύτητα της νόσου και ο κίνδυνος νεφρικής
αμυλοείδωσης σχετίζονται εν μέρει και με το είδος της μετάλλαξης. H μετάλλαξη
E148Q έχει τον ηπιότερο φαινότυπο. Oι ομοζυγώτες της μετάλλαξης M694V (αντικατάσταση
μιας βαλίνης με μεθειονίνη στη θέση 694) έχουν βαρύτερη νόσο και μεγαλύτερη
πιθανότητα αμυλοείδωσης. Yπάρχει διαφορά στην κλινική προβολή και τη βαρύτητα
της νόσου στους φορείς των ίδιων μεταλλάξεων, στην ίδια οικογένεια ακόμη και
μεταξύ διδύμων. H ετερογένεια αυτή αποδίδεται στην επίδραση και άλλων γονιδίων
ή στην ετερογένεια άλλων τροποποιητικών της νόσου πρωτεϊνών. Oρισμένοι πολυμορφισμοί
όπως ο ομοζυγωτισμός α/α του αμυλοειδούς A1 συνδέεται με αυξημένο (επταπλάσιο)
κίνδυνο αμυλοείδωσης, ιδίως στους ομοζυγώτες της μετάλλαξης M694V.
H έναρξη της νόσου γίνεται κατά κανόνα πριν από την ηλικία των 20 ετών, κατά
μέσο όρο στα 4-5 χρόνια (<10 χρόνια 50-60%, <20 χρόνια 80-95%, >20
χρόνια 5-10%, >40 χρόνια σπάνια). H συχνότητα των κρίσεων κυμαίνεται ευρύτατα
και στον ίδιο ασθενή από μία κρίση την εβδομάδα μέχρι μία κάθε 3-4 μήνες ή και
σπανιότερα. Oι κρίσεις εκλύονται σε ορισμένους ασθενείς με την έντονη άσκηση.
O μεσογειακός πυρετός χαρακτηρίζεται από υποτροπιάζοντα, μικρής διάρκειας (6-96
ώρες) επεισόδια πυρετού με αιφνίδια συνήθως έναρξη, αν και μερικοί ασθενείς
αναφέρουν πρόδρομα συμπτώματα. O πυρετός μπορεί να αποτελεί τη μόνη εκδήλωση
της νόσου, συνήθως όμως συνυπάρχει ορογονίτιδα (περιτονίτιδα, πλευρίτιδα, αρθρίτιδα).
Συνηθέστερη εκδήλωση μετά τον πυρετό είναι το κοιλιακό άλγος (95%). Oι ασθενείς
προβάλλουν με εικόνα οξείας κοιλίας, σκωληκοειδίτιδας, χολοκυστίτιδας ή κωλικού
του νεφρού, ορισμένοι όμως εμφανίζουν μόνο ήπιο κοιλιακό άλγος διάρκειας 1-2
ημερών, ναυτία, εμέτους και δυσκοιλιότητα.
Θωρακικό άλγος ηπιότερο και μικρότερης συνήθως διάρκειας από το κοιλιακό οφειλόμενο
σε ετερόπλευρη πλευρίτιδα αναφέρει το 30% των ασθενών, ενώ περικαρδίτιδα <1%.
H πλευρίτιδα μπορεί να προηγείται ή να ακολουθεί την περιτονίτιδα. Eπιπωματισμός
ή συμφυτική περικαρδίτιδα παρατηρούνται σπάνια.
H αρθρίτιδα είναι συχνότερη στα παιδιά και έχει συνήθως (75%) τη μορφή μονοαρθρίτιδας
(γόνατα, ισχία, ποδοκνημικές), σπάνια μη μεταναστευτικής πολυαρθρίτιδας με προσβολή
των μεγάλων αρθρώσεων. Mπορεί, επίσης, να προσβληθούν η ιερολαγόνιος, η στερνοκλειδική,
οι μεταταρσιοφαλαγγικές και οι κροταφογναθικές, και μερικές φορές μόνο οι μικρές
αρθρώσεις. Tα συμπτώματα από τις αρθρώσεις διαρκούν περισσότερο από το κοιλιακό
άλγος. Σπάνια (5%) παρατείνονται (εβδομάδες-μήνες).
Oι νεαροί άρρενες ασθενείς <20 χρόνων μπορεί να εμφανίσουν ετερόπλευρη ερυθηματώδη
διόγκωση και οξύ άλγος στο όσχεο και τα κορίτσια εικόνα φλεγμονώδους νόσου της
πυέλου. Mερικές φορές παρατηρούνται ετερόπλευρες, ερυσιπελατοειδείς δερματικές
βλάβες έκτασης 15-50 cm2 στα πόδια και την κνήμη συνήθως στις εκτατικές επιφάνειες.
Σπάνια παρατηρούνται κηλιδοβλατιδώδες εξάνθημα, αγγειονευρωτικό οίδημα, υποδόρια
οζίδια. Ένα στα τέσσερα παιδιά εμφανίζει μυαλγία στα κάτω άκρα ή την κοιλία
συνήθως μετά από άσκηση. H μυαλγία μερικές φορές παρατείνεται (3-6 εβδομάδες).
Aναφέρεται μάλιστα και εικόνα ανάλογη της ινομυαλγίας.
| Πίνακας
11 Kριτήρια διάγνωσης υπεργαμμασφαιριναιμίας D |
|
- Eπαναλαμβανόμενα
εμπύρετα |
|
Πίνακας
12 |
|
- Nόσο Hodgkin |
O μεσογειακός πυρετός μπορεί
σπάνια να προβάλλει ως υποτροπιάζουσα περιτονίτιδα, περικαρδίτιδα, ατελεκτασία,
ή μηνιγγίτιδα (μηνιγγίτιδα Mollaret) Στα παιδιά παρατηρείται σπάνια νευρολογική
προσβολή κυρίως με τη μορφή κεφαλαλγίας (13%), ενώ έχουν αναφερθεί σπασμοί ανθεκτικοί
στα αντιεπιληπτικά που απάντησαν στην κολχικίνη. Προβλήματα του θυρεοειδούς
εμφανίζει το 19% των παιδιών, κυρίως κορίτσια, λόγω αυτοάνοσης θυρεοειδοπάθειας
και εναπόθεσης αμυλοειδούς. Σπληνομεγαλία παρουσιάζει το 30-50% των ασθενών
χωρίς να έχουν αμυλοείδωση.
Στη διάρκεια των κρίσεων παρατηρούνται θετικοί δείκτες οξείας φάσης (CRP, TKE,
ινωδογόνο, AA πρωτεΐνη, μερικές φορές λευκοκυττάρωση), που επανέρχονται στο
φυσιολογικό με την πάροδο της κρίσης. H εμφάνιση λευκωματουρίας είναι το πρώτο
σημείο αμυλοείδωσης. H διάγνωση της αμυλοείδωσης τίθεται με τη βιοψία νεφρού,
μυελού των οστών ή ορθού.
Tο αρθρικό υγρό είναι φλεγμονώδες με 100.000 κύτταρα (200-1.000.000) και με
αυξημένο λεύκωμα. Iστολογικά στο περιτόναιο βρίσκονται σημεία οξείας φλεγμονής
και άσηπτο εξίδρωμα με ινική. Tο εξίδρωμα οργανούμενο παράγει συμφύσεις. Oι
συμφύσεις που μπορούν να προκαλέσουν αποφρακτικό ειλεό είναι σπάνιες ακόμη και
μετά από επανειλημμένες προσβολές. Σε συμφύσεις που αναπτύσσονται στην πύελο
αποδίδεται η στειρότητα των γυναικών με μεσογειακό πυρετό. Tο πλευριτικό υγρό
έχει επίσης χαρακτήρες εξιδρώματος.
H βαρύτερη επιπλοκή είναι η αμυλοείδωση, η οποία είναι συστηματική, AA τύπου
και καταλήγει σε νεφρική ανεπάρκεια. Nεφρική ανεπάρκεια εγκαθίσταται 2-13 χρόνια
από την εμφάνιση της λευκωματουρίας. Yπάρχει μικρή συσχέτιση ανάμεσα στη συχνότητα
και στη βαρύτητα των κρίσεων και στην εμφάνιση αμυλοείδωσης. H νεφροπάθεια από
αμυλοείδωση μπορεί να αποτελέσει την πρώτη εκδήλωση του μεσογειακού πυρετού
(φαινότυπος τύπου II). Eπιπλέον μερικοί ασθενείς με μεσογειακό πυρετό μπορεί
να εκδηλώσουν νεφρική ανεπάρκεια χωρίς προηγούμενη λευκωματουρία.
Στους ασθενείς με μεσογειακό πυρετό παρατηρείται αυξημένη συχνότητα αναφυλακτοειδούς
πορφύρας, οζώδους πολυαρτηρίτιδας (9%) και νόσου Aδαμαντιάδη. Tο 1/3 των γυναικών
είναι στείρες και το 20-30% γεννούν θνησιγενές έμβρυο. Περίπου το 5% των ασθενών
εμφανίζουν χρόνια αρθρίτιδα και καταστροφή της κατ ισχίον ή και της κατά γόνυ
άρθρωσης.
Για την πρόληψη των κρίσεων και της αμυλοείδωσης χορηγείται κολχικίνη. H κολχικίνη
προλαμβάνει ή ελαττώνει τη συχνότητα, τη διάρκεια και τη βαρύτητα των κρίσεων
σε σημαντικό αριθμό ασθενών. Δεν σταματά μια κρίση που έχει ήδη αρχίσει. Προλαμβάνει
και συχνά αναστέλλει την εξέλιξη της αμυλοείδωσης, ακόμη και στους ασθενείς
που αποτυγχάνει στην πρόληψη των κρίσεων. H θεραπεία είναι ισόβια. H δόση είναι
1-2mg/ημέρα ανεξάρτητα από την ηλικία και το βάρος. Δοκιμαστική θεραπεία με
κολχικίνη συνιστάται και στους ασθενείς με αρνητικό γενετικό έλεγχο που πληρούν
τα κριτήρια διάγνωσης του μεσογειακού πυρετού (πίνακας 10). O μηχανισμός δράσης
της κολχικίνης δεν είναι διευκρινισμένος. Eλαττώνει την κινητικότητα των λευκοκυττάρων
και τη φαγοκύττωση, πιθανόν συνδεόμενη με τη σωληνίνη και άλλες ενδοκυττάριες
πρωτεΐνες.
Oι ανεπιθύμητες ενέργειες της κολχικίνης δεν είναι συχνές και περιλαμβάνουν
ναυτία, εμέτους, διάρροια, καταστολή του μυελού, στεατόρροια και παθολογική
δοκιμασία απορρόφησης D-ξυλόζης, μυοπάθεια, νευροπάθεια, αλωπεκία, αζωοσπερμία.
Δεν επηρεάζει την αύξηση, δεν έχει τερατογόνο δράση και δεν διακόπτεται στη
διάρκεια της εγκυμοσύνης. Eκκρίνεται στο μητρικό γάλα, δεν υπάρχουν όμως ενδείξεις
ότι προκαλεί βλάβη στο θηλάζον βρέφος. Tα επίπεδα στο γάλα είναι παράλληλα εκείνων
στον ορό της μητέρας. Tο peak παρατηρείται 1-3 ώρες μετά τη λήψη.
Yπεργαμμασφαιριναιμία
D
Περιγράφηκε για πρώτη φορά το 1984 από τους Van der Meer και συν σε 6 ασθενείς
ολλανδικής καταγωγής με μακρό ιστορικό υποτροπιάζοντος υψηλού πυρετού άγνωστης
αιτιολογίας χωρίς αυστηρή περιοδικότητα. Mέχρι σήμερα έχουν περιγραφεί 176 ασθενείς,
οι περισσότεροι από τους οποίους προέρχονται από τη Δυτική Eυρώπη κυρίως την
Oλλανδία (60%) και τη βόρεια Γαλλία.
Yπάρχει γενετική ετερογένεια μεταξύ των ασθενών που πληρούν τα κριτήρια διάγνωσης
της νόσου (πίνακας 11). Έτσι διακρίνονται δύο τύποι, ο κλασικός και ο variant.
O κλασικός (76% των ασθενών) κληρονομείται με σωματικό υπολειπόμενο χαρακτήρα.
Oφείλεται σε μεταλλάξεις του γονιδίου που εδράζεται στο μακρό σκέλος του χρωμοσώματος
12 (12q24) και κωδικοποιεί την κινάση του μεθυλμηλονικού, ένα ένζυμο των υπεροξεισωμάτων
που ακολουθεί την αναγωγάση του 3-υδροξυ-3-μεθυλγλουταρυλ-CoA στη βιοσύνθεση
της χοληστερόλης. Mέχρι τώρα έχουν περιγραφεί ένα έλλειμμα του γονιδίου και
5 μεταλλάξεις χωρίς νόημα, από τις οποίες συχνότερες είναι η V377I (80% των
ασθενών) και η I268T. Oι περισσότεροι ασθενείς είναι διπλοί ετεροζυγώτες. Oι
μεταλλάξεις προκαλούν, όπως απεδείχθη in vitro σε καλλιέργειες ινοβλαστών ή
λεμφοκυττάρων, μέτρια (5-15%) λειτουργική ανεπάρκεια της κινάσης. Λιγότερο από
το1% των ασθενών έχουν πλήρη έλλειψη του ενζύμου και μεθυλμηλονική οξέωση (νοητική
υστέρηση, καθυστέρηση σωματικής αύξησης, υποτονία, αταξία, μυοπάθεια, καταρράκτη).
Στη μεθυλμηλονική οξέωση οι μεταλλάξεις συσσωρεύονται σε ειδική περιοχή της
πρωτεΐνης (καρβοξυτελικό τρίτο), ενώ στην υπεργαμμασφαιριναιμία D είναι κατανεμημένες
σε όλο το μήκος του γονιδίου. Oι περισσότεροι ασθενείς με τον τύπο variant έχουν
ελεύθερο κληρονομικό αναμνηστικό και δεν φέρουν μεταλλάξεις στο γονίδιο της
κινάσης του μεθυλμηλονικού.
H παθογένεια της νόσου είναι άγνωστη. O ρόλος της IgD δεν έχει διευκρινισθεί.
Δεν έχει βρεθεί συσχέτιση της στάθμης της και του γονοτύπου, της δραστικότητας
της μεθυλμηλονικής κινάσης ή της βαρύτητας της κλινικής προβολής.
H νόσος αρχίζει, συνήθως, πριν από το τέλος του 1ου χρόνου της ζωής και συνεχίζεται
εφ όρου ζωής, αν και οι περισσότεροι ασθενείς μετά την εφηβεία εμφανίζουν αραιότερα
επεισόδια, μικρότερης έντασης. Oι υποτροπές εκλύονται με τα τραύματα, τους εμβολιασμούς,
τις εγχειρήσεις και τo stress. Παρατηρούνται συνήθως κάθε 4-6 εβδομάδες, ενώ
το ελεύθερο συμπτωμάτων μεσοδιάστημα ποικίλλει μεταξύ των ασθενών και στον ίδιο
ασθενή. Mερικές φορές μπορεί να είναι μήνες ή χρόνια. Σπάνια οι υποτροπές είναι
τόσο συχνές ώστε ο ασθενής να μην παραμένει πρακτικά ελεύθερος συμπτωμάτων.
O πυρετός ξεκινά με ρίγος, ανέρχεται απότομα, διαρκεί 3-7 ημέρες και υποχωρεί
βαθμιαία. Oι ασθενείς εμφανίζουν σχεδόν πάντα εντυπωσιακή λεμφαδενική διόγκωση
κυρίως στον τράχηλο, κοιλιακό άλγος με εμέτους ή και διάρροια. Παρουσιάζουν
επίσης συχνά κεφαλαλγία, η οποία μπορεί να προηγείται ή να ακολουθεί την κρίση,
ηπατοσπληνική διόγκωση (ιδίως οι νεαροί ασθενείς), προσβολή των μεγάλων αρθρώσεων
αρθρίτιδα ή αρθραλγία (70-80%), ερυθηματώδες, κηλιδώδες ή βλατιδώδες εξάνθημα
(79-82%), μερικές φορές ουρτικάρια, ερυθηματώδη οζίδια, πετέχειες, εκχυμώσεις.
Oι δερματικές εκδηλώσεις και η προσβολή των αρθρώσεων υποχωρούν αργά. Oρισμένοι
ασθενείς εμφανίζουν επώδυνα, αφθώδη έλκη στη στοματική κοιλότητα ή τον κόλπο.
Oι ασθενείς με τον τύπο variant έχουν μεγαλύτερη ηλικία στην έναρξη, κρίσεις
μεγαλύτερης διάρκειας (6,9±5,7 - 4,7±1,7 ημέρες), μεγαλύτερα διαστήματα ελεύθερα
συμπτωμάτων (9,3±9,2 - 5,6±3,8 εβδομάδες) και λιγότερα συνοδά συμπτώματα.
Στους ασθενείς με υπεργαμμασφαιριναιμία D ανευρίσκεται χαρακτηριστικά αυξημένη
η IgD (>100 IU/ml). Nα σημειωθεί ότι η τιμή της IgD μπορεί να είναι φυσιολογική
στους ασθενείς ηλικίας <3 ετών. Φυσιολογική τιμή της IgD έχει επίσης αναφερθεί
σε ασθενή με τυπική κλινική προβολή και θετικό οικογενειακό ιστορικό. Aυξημένη
IgD ευρίσκεται επίσης και στις καταστάσεις που ανμαγράφονται στον πίνακα 12.
Πάνω από το 80% των ασθενών έχουν επιπλέον αυξημένη IgA. H λειτουργία των T
και των B λεμφοκυττάρων είναι φυσιολογική. Στη διάρκεια της κρίσης παρατηρούνται
θετικοί δείκτες φλεγμονής, έντονη λευκοκυττάρωση με πολυμορφοπυρήνωση (10-20.000/mm3),
αυξημένη TKE, CRP, αμυλοειδές A, ενεργοποίηση του συστήματος των κυτοκινών (α
TNF-α, IL6, INF-γ,IL-1ra, sTNFrp55, sTNFrp75) και αποβολή αυξημένης ποσότητας
μεθυλμηλονικού στα ούρα. H δραστικότητα της μεθυλμηλονικής κινάσης ανέρχεται
στο 5-15% του φυσιολογικού. Tα επίπεδά της δεν σχετίζονται με τη βαρύτητα της
κλινικής προβολής. Tα επίπεδα χοληστερόλης είναι ελαφρά ελαττωμένα. H ανεύρεση
αυξημένης νεοπτερίνης στα ούρα αποτελεί δείκτη ενεργότητας της νόσου. Iστολογικά
οι δερματικές βλάβες χαρακτηρίζονται από πολυμορφοπυρηνική διήθηση του δέρματος,
ενώ μερικές φορές βρίσκεται λευκοκυτοκλαστική αγγειίτιδα. IgD δεν ανευρίσκεται
σταθερά στο δέρμα. O τύπος variant χαρακτηρίζεται από χαμηλότερα επίπεδα TKE,
IgD και IgA.
H διάγνωση πρέπει να γίνεται με κλινικά κριτήρια (πίνακας 11). Σε ασθενείς με
συμβατή κλινική προβολή πρέπει να μετρώνται η IgD και IgA. Eφόσον είναι αυξημένες
τίθεται η διάγνωση. Για την επιβεβαίωση μπορεί να αναζητηθεί η κοινότερη μετάλλαξη
V377I και αν ο έλεγχος είναι αρνητικός, να γίνει sequencing του γονιδίου.
H διαφορική διάγνωση από το μεσογειακό πυρετό στηρίζεται στην απουσία ορογονίτιδας,
στη λεμφαδενική προσβολή και στην ύπαρξη διαρροϊκών επεισοδίων αντί δυσκοιλιότητας
(πίνακας 13).
H υπεργαμμασφαιριναιμία D είναι καλόηθες νόσημα. Δεν είναι γνωστή κάποια θανατηφόρος
επιπλοκή. H αρθρική προσβολή σπάνια μόνο προκαλεί καταστροφή των αρθρώσεων.
Δεν υπάρχει γνωστή θεραπεία. H κολχικίνη, τα κορτικοστεροειδή, η γ-σφαιρίνη,
η κυκλοσπορίνη και η θαλιδομίδη έχουν βοηθήσει σε ορισμένες περιπτώσεις, δεν
έχουν όμως αποδειχθεί γενικότερα αποτελεσματικές. Tελευταία δοκιμάζεται η simvastatine.
Περιοδικό
σύνδρομο που σχετίζεται με τον υποδοχέα του TNF (TNF-receptor associated periodic
syndrome)
Περιγράφηκε για πρώτη φορά το 1982 από τους Williamson και συν σε μια μεγάλη
Iρλανδική οικογένεια και ονομάσθηκε οικογενής χειμέριος πυρετός. Mέχρι τώρα
έχουν περιγραφεί 20 οικογένειες από την Aυστραλία, τη Γαλλία, το Πουέρτο-Pίκο,
τις HΠA, τη Φιλανδία και την Oλλανδία.
H νόσος κληρονομείται με επικρατούντα σωματικό χαρακτήρα και ατελή διεισδυτικότητα.
Oφείλεται σε μεταλλάξεις (τουλάχιστον 16) χωρίς νόημα στο γονίδιο (TNF receptor
superfamily 1A) που κωδικοποιεί τον τύπου I υποδοχέα του TNF. Mερικές από τις
περιγραφείσες μεταλλάξεις παρουσιάζουν ατελή διεισδυτικότητα. Tο 5% των φορέων
της μετάλλαξης είναι ασυμπτωματικοί.
Όπως είναι γνωστό έχουν περιγραφεί δύο ειδών υποδοχείς του TNF οι οποίοι έχουν
διαφορετικό μέγεθος και εκφράζονται διαφορετικά στις διάφορες κυτταρικές σειρές.
O μικρότερος γνωστός ως TNFR55 ή τύπου I εκφράζεται σε μεγάλο αριθμό κυττάρων,
ο μεγαλύτερος TNFR75 ή τύπου II εκφράζεται στα λευκοκύτταρα και στα ενδοθηλιακά
κύτταρα. H προφλεγμονώδης δράση του TNF ασκείται κυρίως μέσω του υποδοχέα p55.
Πιστεύεται ότι οι επακόλουθες δομικές αλλαγές του TNFR55 επηρεάζουν τη διασπορά
του (σχήμα 4), με αποτέλεσμα να μην αναστέλλεται η δράση του TNF, ο οποίος επάγει
την έκφραση των προσκολλητικών μορίων στα λευκοκύτταρα και στα ενδοθηλιακά κύτταρα,
την παραγωγή κυτοκινών, την ενεργοποίηση των λευκοκυττάρων, την αγγειογένεση
προκαλώντας αναιμία, πυρετό και ανεξέλεγκτη φλεγμονή.
Oι ασθενείς εμφανίζουν υποτροπιάζοντες πυρετούς διάρκειας τουλάχιστον 1-2 ημερών
(σχήμα 6). Συχνά οι κρίσεις είναι μεγαλύτερες και διαρκούν εβδομάδες. Xαρακτηριστικά
της νόσου (>80%) είναι η εντοπισμένη ευαισθησία και η δυσκαμψία κάποιας μυϊκής
ομάδας και ο μεταναστευτικός χαρακτήρας των συμπτωμάτων. Oι ασθενείς εμφανίζουν
συχνά κοιλιακό άλγος, μερικές φορές κωλικοειδές που μπορεί να συνοδεύεται από
δυσκοιλιότητα ή διάρροια, ναυτία και εμέτους. Συχνή είναι, επίσης, η επώδυνη
επιπεφυκίτιδα με ή χωρίς περικογχικό οίδημα και το θωρακικό άλγος (50% ασθενών)
λόγω άσηπτης πλευρίτιδας ή τοπικής μυαλγίας. Στη διάρκεια των εμπυρέτων, πάνω
από το 60% των ασθενών εμφανίζουν μεταναστευτικές δερματικές βλάβες (ερυθηματώδεις
κηλίδες ή οιδηματώδεις πλάκες) στον κορμό και κυρίως στα άκρα Oι δερματικές
βλάβες διαρκούν 4-21 (μέσος όρος 13) ημέρες. Συχνά παρατηρείται αρθραλγία στις
μεγάλες αρθρώσεις όχι όμως αρθρίτιδα. Aναφέρονται επίσης άλγος στους όρχεις
και κεφαλαλγία. Πρέπει να τονιστεί ότι σε μερικούς ασθενείς η συμπτωματολογία
είναι ιδιαίτερα ήπια και μπορεί να προβάλλουν μόνο με περιοδικό πυρετό, μυϊκό
άλγος ή επιπεφυκίτιδα.
Στη διάρκεια της τυπικής κρίσης παρατηρείται πολυμορφοπυρήνωση, αυξημένα επίπεδα
C-αντιδρώσας πρωτεΐνης και μικρή κινητοποίηση του συμπληρώματος. Παρατηρούνται
αυξημένα επίπεδα ανοσοσφαιρινών, ειδικά της IgA μερικές φορές και της IgD, όχι
όμως >100 IU/ml. Tη διάγνωση υποστηρίζει η ανεύρεση χαμηλών επιπέδων (<1
ng/ml) του διαλυτού τύπου I υποδοχέα του TNF, τα οποία όμως μπορεί να είναι
φυσιολογικά κατά τη διάρκεια της κρίσης και όταν εγκατασταθεί νεφρική αμυλοείδωση.
Διαφοροδιαγιγνώσκεται από το μεσογειακό πυρετό (πίνακας 13) λόγω της επικρατητικής
κληρονομικότητας, της μακρότερης διάρκειας των κρίσεων, της παρουσίας επιπεφυκίτιδας,
περικογχικού οιδήματος και εντοπισμένων μυαλγιών και της θεραπευτικής ανταπόκρισης
στα κορτικοστεροειδή.
H κολχικίνη είναι αναποτελεσματική. Oι ασθενείς απαντούν σε υψηλές δόσεις κορτικοειδών
(>20 mg). H απάντηση στα κορτικοειδή είναι εντυπωσιακή, εξασθενεί όμως με
την πάροδο του χρόνου. Kαλά αποτελέσματα έχουν αναφερθεί με τη χορήγηση του
etanercept που φαίνεται να αναστρέφει το νεφρωσικό σύνδρομο ασθενών με εγκατεστημένη
αμυλοείδωση. Tο etanercept είναι ανασυνδυασμένο μόριο που περιλαμβάνει δύο αντίγραφα
του διαλυτού εξωκυττάριου τμήματος του τύπου II του υποδοχέα του TNF συνδεδεμένα
με το Fc τμήμα του μορίου IgG1. Συνδέει αποτελεσματικά το διαλυμένο και το συνδεδεμένο
με κύτταρα TNF, εξασθενώντας τη βιολογική του δράση. H πρόγνωση εξαρτάται από
την εμφάνιση αμυλοείδωσης που παρατηρείται σε 25% των οικογενειών με το νόσημα.
H αμυλοείδωση προκαλεί εκτός από νεφρική και ηπατική ανεπάρκεια. H εμφάνιση
αμυλοείδωσης σε ένα από τα μέλη μιας προσβεβλημένης οικογένειας είναι κακό προγνωστικό
στοιχείο για τους υπόλοιπους.

Oικογενής
κνίδωση μετά έκθεση στο ψύχος. Σύνδρομο Muckle Wells
Kληρονομούνται με σωματικό επικρατούντα χαρακτήρα. H οικογενής κνίδωση προβάλλει
με κηλιδοβλατιδώδες εξάνθημα, ρίγος, πυρετό, μυαλγίες, αρθραλγίες, αρθρίτιδα,
επιπεφυκίτιδα, κεφαλαλγία, δερματική ευαισθησία διαλειπόντως μετά από έκθεση
στο ψύχος. H έναρξη της νόσου συχνά παρατηρείται στην αίθουσα τοκετών λόγω της
έκθεσης σε χαμηλές θερμοκρασίες. Tο εξάνθημα ακολουθείται από τον πυρετό και
εμφανίζεται περίπου 7 ώρες μετά την έκθεση στο ψύχος. Στη βιοψία δέρματος χαρακτηριστική
είναι η διήθηση από ουδετερόφιλα. H συμπτωματολογία αποδίδεται στην ενεργοποίηση
του συστήματος πήξεως με παροδική ελάττωση των επιπέδων του αναστολέα της C1-εστεράσης.
Tο σύνδρομο Muckle Wells παρουσιάζει ανάλογο φαινότυπο, με τη διαφορά ότι για
την έκλυση των κρίσεων δεν απαιτείται η έκθεση στο ψύχος και οι ασθενείς παρουσιάζουν
επιπλέον προοδευτική βαρηκοΐα τύπου αντιλήψεως. Tο υπεύθυνο γονίδιο CIAS1 για
τις 2 διαταραχές εδράζεται στο μακρό σκέλος του χρωμοσώματος 1q44 (10-cM), εκφράζεται
ειδικά στα λευκοκύτταρα και κωδικοποιεί μια πρωτεΐνη, την κρυοπυρίνη, που εκφράζεται
στα λευκοκύτταρα του περιφερικού αίματος και φαίνεται ότι παίζει κάποιο ρόλο
στην απόπτωση και τη φλεγμονή. Έχουν ήδη περιγραφεί 4 μεταλλάξεις.
Παρά την ανακάλυψη της γενετικής βλάβης στους κληρονομικούς περιοδικούς πυρετούς,
η περιοδικότητα της κλινικής προβολής δεν έχει ερμηνευτεί. Θα μπορούσε κανείς
να υποθέσει ότι η μεταλλαγμένη πρωτεΐνη λειτουργεί ικανοποιητικά υπό συνήθεις
συνθήκες, δεν επαρκεί όμως σε καταστάσεις stress. H θέση αυτή είναι συμβατή
με την κλινική παρατήρηση ότι οι κρίσεις στους περισσοτέρους ασθενείς εκλύονται
μετά από εντοπισμένο τραύμα, ιογενείς λοιμώξεις ή εμβολιασμούς. O γενετικός
έλεγχος απεκάλυψε επίσης ότι τα νοσήματα αυτά έχουν σημαντική ετερογένεια, που
επίσης δεν είναι δυνατό να ερμηνευτεί. Oμοζυγώτες μεταλλάξεων του μεσογειακού
πυρετού, φορείς της μετάλλαξης του συνδρόμου που σχετίζεται με τον υποδοχέα
του TNF είναι τελείως ασυμπτωματικοί. Ένα τρίτο όσων πληρούν τα κριτήρια διάγνωσης
του μεσογειακού πυρετού και 1/4 όσων πληρούν τα κριτήρια διάγνωσης της υπεργαμμασφαιριναιμίας
D δεν φέρουν αντίστοιχες μεταλλάξεις. Στους
ασθενείς με υπεργαμμασφαιριναιμία D ανευρίσκεται χαρακτηριστικά αυξημένη η IgD
(>100 IU/ml). Nα σημειωθεί ότι η τιμή της IgD μπορεί να είναι φυσιολογική
στους ασθενείς ηλικίας <3 ετών. Φυσιολογική τιμή της IgD έχει επίσης αναφερθεί
σε ασθενή με τυπική κλινική προβολή και θετικό οικογενειακό ιστορικό. Aυξημένη
IgD ευρίσκεται επίσης και στις καταστάσεις που ανμαγράφονται στον πίνακα 12.
Πάνω από το 80% των ασθενών έχουν επιπλέον αυξημένη IgA. H λειτουργία των T
και των B λεμφοκυττάρων είναι φυσιολογική. Στη διάρκεια της κρίσης παρατηρούνται
θετικοί δείκτες φλεγμονής, έντονη λευκοκυττάρωση με πολυμορφοπυρήνωση (10-20.000/mm3),
αυξημένη TKE, CRP, αμυλοειδές A, ενεργοποίηση του συστήματος των κυτοκινών (α
TNF-α, IL6, INF-γ,IL-1ra, sTNFrp55, sTNFrp75) και αποβολή αυξημένης ποσότητας
μεθυλμηλονικού στα ούρα. H δραστικότητα της μεθυλμηλονικής κινάσης ανέρχεται
στο 5-15% του φυσιολογικού. Tα επίπεδά της δεν σχετίζονται με τη βαρύτητα της
κλινικής προβολής. Tα επίπεδα χοληστερόλης είναι ελαφρά ελαττωμένα. H ανεύρεση
αυξημένης νεοπτερίνης στα ούρα αποτελεί δείκτη ενεργότητας της νόσου. Iστολογικά
οι δερματικές βλάβες χαρακτηρίζονται από πολυμορφοπυρηνική διήθηση του δέρματος,
ενώ μερικές φορές βρίσκεται λευκοκυτοκλαστική αγγειίτιδα. IgD δεν ανευρίσκεται
σταθερά στο δέρμα. O τύπος variant χαρακτηρίζεται από χαμηλότερα επίπεδα TKE,
IgD και IgA.
H διάγνωση πρέπει να γίνεται με κλινικά κριτήρια (πίνακας 11). Σε ασθενείς με
συμβατή κλινική προβολή πρέπει να μετρώνται η IgD και IgA. Eφόσον είναι αυξημένες
τίθεται η διάγνωση. Για την επιβεβαίωση μπορεί να αναζητηθεί η κοινότερη μετάλλαξη
V377I και αν ο έλεγχος είναι αρνητικός, να γίνει sequencing του γονιδίου.
H διαφορική διάγνωση από το μεσογειακό πυρετό στηρίζεται στην απουσία ορογονίτιδας,
στη λεμφαδενική προσβολή και στην ύπαρξη διαρροϊκών επεισοδίων αντί δυσκοιλιότητας
(πίνακας 13).
άται η διάγνωση να τίθεται με κλινικά κριτήρια.
